
Реферат гемофилия список литературы
Обновлено: 04.07.2024
Гемофилия включает ряд наследственных нарушений свертываемости крови, которые имеют общую эндогенную природу коагуляционного расстройства. Эти нарушения могут быть подразделены на гемофилию А (классическая гемофилия), гемофилию В (болезнь Кристмаса) и болезнь фон Виллебранда.
1. Общая информация и клинические синдромы
Классическая гемофилия -- это дефицит нормального функционального фактора VIII, болезнь Кристмаса -- дефицит нормального фактора IX, а болезнь фон Виллебранда -- недостаток как антигенного, так и функционального фактора VIII, а также ко-фактора этого фактора VIII фон Виллебранда. Факторы внутреннего пути коагуляции определяются по ЧТВ (частичное тромбиновое время). Факторы VIII и IX являются уникальными и чрезвычайно важными компонентами внутреннего пути, и их недостаток сопровождается увеличением ЧТВ. Другие факторы, уникальные для внутреннего пути, а именно факторы XII и XI, также могут генетически отсутствовать, однако это бывает гораздо реже и проявляется клинически значительно мягче.
Определение тромбинового времени характеризует в основном экзогенный путь коагуляции, где фактор VII является основополагающим. Совершенно очевидно, что некоторые факторы, такие как X, V, II и I являются общими для эндогенного и экзогенного путей коагуляции. Таким образом, недостаток факторов VIII или IX обусловливает увеличение ЧТВ при нормальном ПВ. Это отличительный признак гемофилии. Очевидно, что идентификация дефицитного фактора совершенно необходима для определения типа наблюдаемой гемофилии. Его идентификация влияет также на выбор специфического лечения.
2. Генетические типы
Классическая гемофилия (гемофилия А)
Гемофилия А -- это связанное с полом наследственное заболевание, обусловливающее дефицит VIII фактора свертывания. В зависимости от уровня фактора VIII в крови его дефицит часто определяется как тяжелый (менее 2 % его нормальной активности), умеренный (активность составляет 2--5 %) и небольшой (5--30 % активности). Лишь около 50 % больных с тяжелой формой гемофилии имеют семейный анамнез заболевания.
Болезнь Кристмаса (гемофилия В)
Гемофилия В -- это также связанное с полом наследственное заболевание, приводящее к недостаточной активности IX фактора свертывания. Различают тяжелую, умеренную и мягко выраженную формы заболевания. Более 75 % больных имеют семейный анамнез гемофилии. Данное заболевание клинически неотличимо от гемофилии А.
Болезнь фон Виллебранда
Это аутосомно-наследуемое заболевание, приводящее к недостаточной активности фактора VIII в коагуляции (VIII-C), иммунологической активности [VIII-антиген(аг)] и VIII фактора фон Виллебранда (VIII-фВФ). Последний -- это фактор, корригирующий дисфункцию тромбоцитов, которая обнаруживается при болезни фон Виллебранда.
Заболевание наследуется по аутосомно-доминантному типу и имеет вариабельные клинические проявления.
3. Клинические синдромы гемофилии
Слабовыраженная форма заболевания
Эта форма заболевания может оставаться недиагностированной до взрослого возраста, хотя в анамнезе у подобных больных очень часто имеются указания на незначительные, но повторяющиеся эпизоды кровотечения в ранние годы жизни. Сильное кровотечение после небольшой травмы наблюдается редко, но оно может возникнуть вследствие удаления зуба, при тонзилэктомии и других хирургических вмешательствах или после тяжелой травмы. У больных женского пола отмечается легкое образование синяков, что вызывает определенные косметологические трудности, а также обильные менструации. Больные с небольшим дефицитом фактора VIII или IX иногда могут иметь нормальное ЧТВ.
Умеренно выраженные и тяжелые формы заболевания
Эти формы заболевания обычно наблюдаются в младенческом возрасте или в раннем детстве. Заболевание может обнаружиться после циркумцизии. У младенцев, начинающих ходить, случайное падение может вызвать образование повторных и резко выраженных внутримышечных гематом, а прикусывание губ или языка приводит к сильному кровотечению. Сильное кровотечение могут спровоцировать и стоматологические процедуры. У подростков и у взрослых возможно возникновение гематурии (спонтанное или вследствие физического напряжения). Характерные гемартрозы и глубокие мышечные гематомы наблюдаются уже тогда, когда дети свободно ходят и бегают; их наибольшая частота отмечается у детей школьного возраста и у подростков.
Мышечная гематома проявляется болезненностью и напряжением прежде чем появятся такие кардинальные симптомы, как припухлость и повышение местной температуры. Ретроперитонеальную гематому не всегда легко распознать; больной может жаловаться на боль в паху, в месте прикрепления поясничных мышц вследствие кровоизлияния в фасциальное пространство.
Кровоизлияния в полость сустава обычно распознаются по ощущению распирания в нем и по ограничению его подвижности. Появляются боль и припухлость в области сустава. Наиболее часто (в убывающем порядке) поражаются коленные, локтевые, голеностопные, бедренные и плечевые суставы. В случае несвоевременного лечения и, следовательно, замедленного рассасывания крови может иметь место хроническая артропатия. Повторные кровотечения в полость сустава ведут к гипертрофии синовиальной оболочки, хроническому воспалению с образованием фиброзных сращений и анкилозированием сустава. Может наблюдаться тяжелая деформация сустава.
4. Лечение
Заместительная терапия
Важное значение для выбора адекватного лечения имеет специфическая идентификация дефицитного фактора свертывания. В ситуации, не позволяющей установить специфическую аномалию, может использоваться свежезамороженная плазма, содержащая все факторы свертывания. Однако для достижения адекватного гемостаза в таких случаях обычно требуются значительные количества плазмы, так что может возникнуть проблема с перегрузкой объемом. Среднему больному необходимо примерно 40 мл плазмы на 1 кг массы тела. В 1 мл нормальной плазмы содержится 1 ЕД факторной активности. Следовательно, переливание плазмы из расчета 40 мл/кг повышает уровень фактора VIII в крови на 100 %. Врач ОНП должен уметь рассчитывать количество вводимого фактора, которое требуется для повышения активности данного фактора в крови больного до желаемого уровня. В зависимости от интенсивности кровотечения (слабое или сильное) врач может стремиться к повышению плазменного уровня данного фактора до 50--100 % рассчитанной нормы. Поскольку период полураспада антигемофильных факторов составляет всего лишь 12 часов, больному может потребоваться повторное переливание в зависимости от локализации, выраженности и длительности кровотечения.
При лечении болезни фон Виллебранда может использоваться и свежезамороженная плазма или криопреципитат. Поскольку одна порция плазмы, получается, от одного донора, риск заражения гепатитом или СПИДом при ее переливании меньше, чем при использовании суммарных кровяных концентратов. Плазма может применяться также у больных с небольшим дефицитом фактора VIII или IX. Ее применение ограничивается риском перегрузки объемом, поскольку безопасным может быть единовременное введение лишь 10--15 мл/кг с ожидаемым повышением активности фактора свертывания на 20-30 %.
До последнего времени криопреципитат был средством выбора при лечении большинства больных с нетяжелой классической гемофилией (5--30 % активность фактора VIII) и пациентов с болезнью фон Виллебранда. Ввиду возможного заражения вирусной инфекцией при переливании кровезаменителей были разработаны альтернативные методы лечения. Применение D-амино-8, D-аргининвазопрессин (DDAVP) из расчета 0,3 мкг на 1 кг массы тела почти всегда позволяет повысить активность VIII фактора до уровня, обеспечивающего гемостаз у таких больных. Этот препарат является аналогом вазопрессина и, следовательно, не ассоциируется с передачей инфекционного заболевания. К сожалению, DDAVP неэффективен при тяжелой гемофилии и в большинстве случаев умеренно выраженной гемофилии, а также при тяжелой болезни фон Виллебранда и при ее форме ИВ. Последняя является необычной формой болезни Виллебранда, поэтому ее диагностика требует тщательного обследования больного специалистом-гематологом.
Использование гемоконцентратов сделало лечение гемофилии более доступным для повседневной практики. Доза концентрата должна соответствовать тяжести кровотечения. При небольшом кровотечении может быть вполне достаточной однократная инфузия нескольких порций криопреципитата в ОНП. Больной предупреждается о необходимости повторного обращения в ОНП при возобновлении кровотечения. Криопреципитат содержит большое количество фактора VIII -- примерно от 5 до 10 ЕД его активности на 1 мл. Порция криопреципитата, полученного от одного донора, содержит 10 мл общего объема, т. е. 50--100 ЕД активности фактора VIII на 1 пакет криопреципитата.
Обработанные нагреванием концентраты VIII фактора в настоящее время доступны в различных лечебных учреждениях общего профиля. Эти гемоконцентраты приготавливаются из депонированной плазмы, лиофилизированной, а затем разбавленной стерильной водой до необходимого объема. Суммарная активность указана на упаковке. Основное преимущество гемоконцентрата состоит в том, что при его использовании не происходит перегрузки объемом; но поскольку этот продукт получается из депонированной плазмы, его введение сопряжено с большим риском заражения гепатитом и СПИДом. Как полагают, обработка препарата нагреванием инактивирует ВИЧ (HIV/ HTLV-Ш), возбудитель СПИДа. Тем не менее, тестирование крови на ВИЧ и на антитела к нему проводится у всех доноров, Концентраты являются более дорогостоящими препаратами. Число единиц активности на 1 мл может значительно варьировать в различных препаратах.
Лиофилизированные концентраты факторов II, VII, IX и X (протромбиновый комплекс) используются, прежде всего, для лечения дефицита фактора IX. По техническим причинам этот комплекс не сепарируется, поэтому его использование сопряжено с более высоким риском заражения гепатитом и СПИДом, а также возникновения тромботических осложнений. Средняя активность фактора IX в концентрате составляет 20 ЕД/мл.
Дополнительные мероприятия
Мышечная гематома и подкожное кровоизлияние могут быть уменьшены с помощью различных местных воздействий (давящая повязка, применение льда, иммобилизация посредством эластичного бинта). Гемартрозы обычно наблюдаются лишь при тяжелой гемофилии и практически никогда -- при болезни фон Виллебранда. Временная иммобилизация сустава с помощью шинирования предупреждает его дальнейшее травмирование, ограничивает припухание и устраняет боль.
Важное значение при гемофилии имеет адекватная аналгезия. Все большее число анальгетиков содержит ацетилсалициловую кислоту; следует воздержаться от их использования при гемофилии, так как они тормозят агрегацию тромбоцитов и усугубляют гемостатические осложнения. В качестве альтернативных препаратов могут использоваться пропоксифен, ацетаминофен и кодеин. Следует избегать применения внутримышечных инъекций.
Противовоспалительные препараты (в особенности кортикостероиды) применяются при гематурии, остром гемартрозе и хроническом синовиите. Их доза обычно составляет 1 мг/кг в день.
Специальные указания
В отдельных случаях больные с тяжелой гемофилией не поддаются соответствующей заместительной терапии. Такие больные могут составить небольшую, но терапевтически сложную группу пациентов с циркулирующими антикоагулянтами (ингибиторы). При лечении этого серьезного осложнения необходима консультативная помощь гематолога.
1. Болезни крови у пожилых: Пер. с англ. / Под ред. Б79 М. Дж. Денхэма, И. Чанарина. -- М.: Медицина, 1989.
2. Неотложная медицинская помощь: Пер. с англ./Под Н52 ред. Дж. Э. Тинтиналли, Р.Л. Кроума, Э. Руиза. -- М.: Медицина, 2001.
2. Момот А.П. Принципы и алгоритмы клинико-лабораторной диагностики. СПб. 2006.
3. Протокол ведения больных "Гемофилия". Проблемы стандартизации в здравоохранении 2006, с. 18 - 74
5. BIV Astermark J, Petrini P, Tengborn L, et al. Primary prophylaxis in severe haemophilia should be started at an early age but can be individualized. Br J Haematol 1999; 105: 1109-13
6. Canadian Hemophilia Standards Group. Canadian Comprehensive Care Standards for Hemophilia and Other Inherited Bleeding Disorders, First Edition, June 2007
7. Castaman G, Mancuso ME, Giacomelli SH, et al. Molecular and phenotypic determinants of the response to desmopressin in adult patients with mild hemophilia A. J Thromb Haemost 2009; 7(11): 1824-31
8. Collins Peter W, Chalmers E, Daniel P. Hart DP, at al. Diagnosis and treatment of. factor VIII and IX inhibitors in congenital haemophilia: (4th edition). British Journal oФ. Haematology. Volume 160, Issue 2, pages 153 - 170, January 2013
9. Colvin BT, Astermark J, Fischer K, Gringeri A, Lassila R, Schramm W, Thomas A, Ingerslev J; Inter Disciplinary Working Group. European principles oФ. haemophilia care. Haemophilia 2008; 14(2): 361-74
10. De Moerloose P, Fischer K, Lambert T et al. Recommendations for assessment, monitoring and follow-up of patients with haemophilia. Haemophilia 2012; 18: 319-25
11. Fonseca, A., Nagel, K., Decker, K., Pukulakatt, M., Pai, M., Walton, M., & Chan, A. K. C. (2016). Central venous access device insertion and perioperative management of patients with severe haemophilia A: a local experience. Blood Coagulation & Fibrinolysis, 27(2), 156 - 159
12. Franchini M, Zaffanello M, Lippi G. The use of desmopressin in mild hemophilia A. Blood Coagul fibrinolysis 2010; 21(7): 615-9
13. Johnsen, Jill M. et al "Novel approach to genetic analysis and results in 3000 hemophilia patients enrolled in the My Life, Our Future initiative." Blood Advances 1.13 (2017): 824 - 834
14. Golestani M, Eshghi P, Rasekh HR, Cheraghali AM, Salamzadeh J, Imani A. Comparison of bypassing agents in bleeding reduction in treatment of bleeding episodes in patients with haemophilia and inhibitors. Iran Red Crescent Med J. 2014; 16(12): e24551
15. Gouw, Samantha C. et al "F8 gene mutation type and inhibitor development in patients with severe hemophilia A: systematic review and meta-analysis." Blood 119.12 (2012): 2922 - 2934
17. Mannucci P.M., Garagiola I. Factor VIII products in haemophilia A: one size fits all? Thromb Haemost 2015; 113(05): 911 - 914
18. Nilsson IM, Berntorp E, 19. Oldenburg J, Mahlangu JN, Kim B, Schmitt C, Callghan MU, Young G, Santagostino E, Kruse-Jarres R, Negrier C, Kessler C, Valente N, Asikanius E, Levy GG, Windyga J, Shima M. Emicizumab Prophylaxis in Hemophilia A with Inhibitors. N Engl J Med (2017) 377 (9)809 - 818
20. Richards M, Williams M, Chalmers E, at al. A United Kingdom Haemophilia Centre Doctors' Organization guideline approved by the British Committee for Standards in Haematology: guideline on the use of prophylactic factor VIII concentrate in children and adults with severe haemophilia A. British Journal of Haematology. Volume 149, Issue 4, pages 498 - 507, May 2010
21. Srivastava A, et al. Guidelines for the management of hemophilia (WFH). Haemophilia 2013: 19; e1 - e47
22. Stonebraker JS, Bolton-Maggs PH, Soucie JM, Walker I, Brooker M. A study of variations in the reported haemophilia A prevalence around the world. Haemophilia 2010; 16(1): 20 - 32
23. Pediatrics Nov Ullman A.J., Marsh N., Mihala G. et al. Complications of Central Venous Access Devices: A Systematic Review. 2015, 136 (5) e1331 - e1344
24. Valentino, L., Ewenstein, B., Navickis, R. J., & Wilkes, M. M. (2004). Central venous access devices in haemophilia. Haemophilia, 10(2), 134-46
Если у вас возникли сложности с курсовой, контрольной, дипломной, рефератом, отчетом по практике, научно-исследовательской и любой другой работой - мы готовы помочь.


Министерство образования Российской Федерации
Пензенский государственный университет
Медицинский институт
Кафедра терапии
Курс гематологии
Зав. кафедрой д.м.н.
Реферат на тему:
Гемофилия
Выполнила: студентка V курса
Проверил: к.м.н., доцент
Пенза 2008
План
Введение
1.Общая информация о гемофилии
2. Генетические типы
Классическая гемофилия (гемофилия А)
Болезнь Кристмаса (гемофилия В)
Болезнь фон Виллебранда
3. Клинические синдромы гемофилии
Слабовыраженная форма заболевания
Умеренно выраженные и тяжелые формы заболевания
4. Лечение
Заместительная терапия
Дополнительные мероприятия
Специальные указания
Литература
Введение
Гемофилия включает ряд наследственных нарушений свертываемости крови, которые имеют общую эндогенную природу коагуляционного расстройства. Эти нарушения могут быть подразделены на гемофилию А (классическая гемофилия), гемофилию В (болезнь Кристмаса) и болезнь фон Виллебранда.
1. Общая информация и клинические синдромы
Классическая гемофилия — это дефицит нормального функционального фактора VIII, болезнь Кристмаса — дефицит нормального фактора IX, а болезнь фон Виллебранда — недостаток как антигенного, так и функционального фактора VIII, а также ко-фактора этого фактора VIII фон Виллебранда. Факторы внутреннего пути коагуляции определяются по ЧТВ (частичное тромбиновое время). Факторы VIII и IX являются уникальными и чрезвычайно важными компонентами внутреннего пути, и их недостаток сопровождается увеличением ЧТВ. Другие факторы, уникальные для внутреннего пути, а именно факторы XII и XI, также могут генетически отсутствовать, однако это бывает гораздо реже и проявляется клинически значительно мягче.
Определение тромбинового времени характеризует в основном экзогенный путь коагуляции, где фактор VII является основополагающим. Совершенно очевидно, что некоторые факторы, такие как X, V, II и I являются общими для эндогенного и экзогенного путей коагуляции. Таким образом, недостаток факторов VIII или IX обусловливает увеличение ЧТВ при нормальном ПВ. Это отличительный признак гемофилии. Очевидно, что идентификация дефицитного фактора совершенно необходима для определения типа наблюдаемой гемофилии. Его идентификация влияет также на выбор специфического лечения.
2. Генетические типы
Классическая гемофилия (гемофилия А)
Гемофилия А — это связанное с полом наследственное заболевание, обусловливающее дефицит VIII фактора свертывания. В зависимости от уровня фактора VIII в крови его дефицит часто определяется как тяжелый (менее 2 % его нормальной активности), умеренный (активность составляет 2—5 %) и небольшой (5—30 % активности). Лишь около 50 % больных с тяжелой формой гемофилии имеют семейный анамнез заболевания.
Болезнь Кристмаса (гемофилия В)
Гемофилия В — это также связанное с полом наследственное заболевание, приводящее к недостаточной активности IX фактора свертывания. Различают тяжелую, умеренную и мягко выраженную формы заболевания. Более 75 % больных имеют семейный анамнез гемофилии. Данное заболевание клинически неотличимо от гемофилии А.
Болезнь фон Виллебранда
Это аутосомно-наследуемое заболевание, приводящее к недостаточной активности фактора VIII в коагуляции (VIII-C), иммунологической активности [VIII-антиген(аг)] и VIII фактора фон Виллебранда (VIII-фВФ). Последний — это фактор, корригирующий дисфункцию тромбоцитов, которая обнаруживается при болезни фон Виллебранда.
Заболевание наследуется по аутосомно-доминантному типу и имеет вариабельные клинические проявления.
3. Клинические синдромы гемофилии
Слабовыраженная форма заболевания
Эта форма заболевания может оставаться недиагностированной до взрослого возраста, хотя в анамнезе у подобных больных очень часто имеются указания на незначительные, но повторяющиеся эпизоды кровотечения в ранние годы жизни. Сильное кровотечение после небольшой травмы наблюдается редко, но оно может возникнуть вследствие удаления зуба, при тонзилэктомии и других хирургических вмешательствах или после тяжелой травмы. У больных женского пола отмечается легкое образование синяков, что вызывает определенные косметологические трудности, а также обильные менструации. Больные с небольшим дефицитом фактора VIII или IX иногда могут иметь нормальное ЧТВ.
Умеренно выраженные и тяжелые формы заболевания
Эти формы заболевания обычно наблюдаются в младенческом возрасте или в раннем детстве. Заболевание может обнаружиться после циркумцизии. У младенцев, начинающих ходить, случайное падение может вызвать образование повторных и резко выраженных внутримышечных гематом, а прикусывание губ или языка приводит к сильному кровотечению. Сильное кровотечение могут спровоцировать и стоматологические процедуры. У подростков и у взрослых возможно возникновение гематурии (спонтанное или вследствие физического напряжения). Характерные гемартрозы и глубокие мышечные гематомы наблюдаются уже тогда, когда дети свободно ходят и бегают; их наибольшая частота отмечается у детей школьного возраста и у подростков.
Мышечная гематома проявляется болезненностью и напряжением прежде чем появятся такие кардинальные симптомы, как припухлость и повышение местной температуры. Ретроперитонеальную гематому не всегда легко распознать; больной может жаловаться на боль в паху, в месте прикрепления поясничных мышц вследствие кровоизлияния в фасциальное пространство.
Кровоизлияния в полость сустава обычно распознаются по ощущению распирания в нем и по ограничению его подвижности. Появляются боль и припухлость в области сустава. Наиболее часто (в убывающем порядке) поражаются коленные, локтевые, голеностопные, бедренные и плечевые суставы. В случае несвоевременного лечения и, следовательно, замедленного рассасывания крови может иметь место хроническая артропатия. Повторные кровотечения в полость сустава ведут к гипертрофии синовиальной оболочки, хроническому воспалению с образованием фиброзных сращений и анкилозированием сустава. Может наблюдаться тяжелая деформация сустава.
4. Лечение
Заместительная терапия
Важное значение для выбора адекватного лечения имеет специфическая идентификация дефицитного фактора свертывания. В ситуации, не позволяющей установить специфическую аномалию, может использоваться свежезамороженная плазма, содержащая все факторы свертывания. Однако для достижения адекватного гемостаза в таких случаях обычно требуются значительные количества плазмы, так что может возникнуть проблема с перегрузкой объемом. Среднему больному необходимо примерно 40 мл плазмы на 1 кг массы тела. В 1 мл нормальной плазмы содержится 1 ЕД факторной активности. Следовательно, переливание плазмы из расчета 40 мл/кг повышает уровень фактора VIII в крови на 100 %. Врач ОНП должен уметь рассчитывать количество вводимого фактора, которое требуется для повышения активности данного фактора в крови больного до желаемого уровня. В зависимости от интенсивности кровотечения (слабое или сильное) врач может стремиться к повышению плазменного уровня данного фактора до 50—100 % рассчитанной нормы. Поскольку период полураспада антигемофильных факторов составляет всего лишь 12 часов, больному может потребоваться повторное переливание в зависимости от локализации, выраженности и длительности кровотечения.
При лечении болезни фон Виллебранда может использоваться и свежезамороженная плазма или криопреципитат. Поскольку одна порция плазмы, получается, от одного донора, риск заражения гепатитом или СПИДом при ее переливании меньше, чем при использовании суммарных кровяных концентратов. Плазма может применяться также у больных с небольшим дефицитом фактора VIII или IX. Ее применение ограничивается риском перегрузки объемом, поскольку безопасным может быть единовременное введение лишь 10—15 мл/кг с ожидаемым повышением активности фактора свертывания на 20-30 %.
До последнего времени криопреципитат был средством выбора при лечении большинства больных с нетяжелой классической гемофилией (5—30 % активность фактора VIII) и пациентов с болезнью фон Виллебранда. Ввиду возможного заражения вирусной инфекцией при переливании кровезаменителей были разработаны альтернативные методы лечения. Применение D-амино-8, D-аргининвазопрессин (DDAVP) из расчета 0,3 мкг на 1 кг массы тела почти всегда позволяет повысить активность VIII фактора до уровня, обеспечивающего гемостаз у таких больных. Этот препарат является аналогом вазопрессина и, следовательно, не ассоциируется с передачей инфекционного заболевания. К сожалению, DDAVP неэффективен при тяжелой гемофилии и в большинстве случаев умеренно выраженной гемофилии, а также при тяжелой болезни фон Виллебранда и при ее форме ИВ. Последняя является необычной формой болезни Виллебранда, поэтому ее диагностика требует тщательного обследования больного специалистом-гематологом.
Использование гемоконцентратов сделало лечение гемофилии более доступным для повседневной практики. Доза концентрата должна соответствовать тяжести кровотечения. При небольшом кровотечении может быть вполне достаточной однократная инфузия нескольких порций криопреципитата в ОНП. Больной предупреждается о необходимости повторного обращения в ОНП при возобновлении кровотечения. Криопреципитат содержит большое количество фактора VIII — примерно от 5 до 10 ЕД его активности на 1 мл. Порция криопреципитата, полученного от одного донора, содержит 10 мл общего объема, т. е. 50—100 ЕД активности фактора VIII на 1 пакет криопреципитата.
Обработанные нагреванием концентраты VIII фактора в настоящее время доступны в различных лечебных учреждениях общего профиля. Эти гемоконцентраты приготавливаются из депонированной плазмы, лиофилизированной, а затем разбавленной стерильной водой до необходимого объема. Суммарная активность указана на упаковке. Основное преимущество гемоконцентрата состоит в том, что при его использовании не происходит перегрузки объемом; но поскольку этот продукт получается из депонированной плазмы, его введение сопряжено с большим риском заражения гепатитом и СПИДом. Как полагают, обработка препарата нагреванием инактивирует ВИЧ (HIV/ HTLV-Ш), возбудитель СПИДа. Тем не менее, тестирование крови на ВИЧ и на антитела к нему проводится у всех доноров, Концентраты являются более дорогостоящими препаратами. Число единиц активности на 1 мл может значительно варьировать в различных препаратах.
Лиофилизированные концентраты факторов II, VII, IX и X (протромбиновый комплекс) используются, прежде всего, для лечения дефицита фактора IX. По техническим причинам этот комплекс не сепарируется, поэтому его использование сопряжено с более высоким риском заражения гепатитом и СПИДом, а также возникновения тромботических осложнений. Средняя активность фактора IX в концентрате составляет 20 ЕД/мл.
Дополнительные мероприятия
Мышечная гематома и подкожное кровоизлияние могут быть уменьшены с помощью различных местных воздействий (давящая повязка, применение льда, иммобилизация посредством эластичного бинта). Гемартрозы обычно наблюдаются лишь при тяжелой гемофилии и практически никогда — при болезни фон Виллебранда. Временная иммобилизация сустава с помощью шинирования предупреждает его дальнейшее травмирование, ограничивает припухание и устраняет боль.
Важное значение при гемофилии имеет адекватная аналгезия. Все большее число анальгетиков содержит ацетилсалициловую кислоту; следует воздержаться от их использования при гемофилии, так как они тормозят агрегацию тромбоцитов и усугубляют гемостатические осложнения. В качестве альтернативных препаратов могут использоваться пропоксифен, ацетаминофен и кодеин. Следует избегать применения внутримышечных инъекций.
Противовоспалительные препараты (в особенности кортикостероиды) применяются при гематурии, остром гемартрозе и хроническом синовиите. Их доза обычно составляет 1 мг/кг в день.
Специальные указания
В отдельных случаях больные с тяжелой гемофилией не поддаются соответствующей заместительной терапии. Такие больные могут составить небольшую, но терапевтически сложную группу пациентов с циркулирующими антикоагулянтами (ингибиторы). При лечении этого серьезного осложнения необходима консультативная помощь гематолога.
Литература
1. Болезни крови у пожилых: Пер. с англ. / Под ред. Б79 М. Дж. Денхэма, И. Чанарина. — М.: Медицина, 1989.
2. Неотложная медицинская помощь: Пер. с англ./Под Н52 ред. Дж. Э. Тинтиналли, Р.Л. Кроума, Э. Руиза. — М.: Медицина, 2001.
Гемофилия – наследственная патология системы гемостаза, в основе которой лежит снижение или нарушение синтеза VIII, IX или XI факторов свертывания крови. Специфическим проявлением гемофилии служит склонность больного к различным кровотечениям: гемартрозам, внутримышечным и забрюшинным гематомам, гематурии, желудочно-кишечным кровотечениям, длительным кровотечениям при операциях и травмах и др. В диагностике гемофилии первостепенное значение имеет генетическое консультирование, определение уровня активности факторов свертываемости, ДНК-исследование, анализ коагулограммы. Лечение гемофилии предполагает проведение заместительной терапии: трансфузии гемоконцентратов с факторами свертывания VIII или IX, свежезамороженной плазмы, антигемофильного глобулина и др.
Общие сведения
Гемофилия – заболевание из группы наследственных коагулопатий, обусловленное дефицитом факторов свертывания плазмы крови и характеризующееся повышенной склонностью к геморрагиям. Распространенность гемофилии А и В составляет 1 случай на 10000-50000 представителей мужского пола. Чаще всего дебют заболевания приходится на ранний детский возраст, поэтому гемофилия у ребенка является актуальной проблемой педиатрии и детской гематологии. Кроме гемофилии, у детей встречаются и другие наследственные геморрагические диатезы: геморрагическая телеангиэктазия, тромбоцитопатия, болезнь Гланцмана т. п.
Причины гемофилии
Гены, обусловливающие развитие гемофилии, сцеплены с половой Х-хромосомой, поэтому заболевание наследуется по рецессивному признаку по женской линии. Наследственной гемофилией болеют практически исключительно лица мужского пола. Женщины являются проводниками (кондукторами, носителями) гена гемофилии, передающими заболевание части своих сыновей.
У здорового мужчины и женщины-кондуктора с одинаковой вероятностью могут родиться как больные, так и здоровые сыновья. От брака мужчины, больного гемофилией со здоровой женщиной рождаются здоровые сыновья или дочери-кондукторы. Описаны единичные случаи гемофилии у девочек, рожденных от матери-носителя и больного гемофилией отца.
Врожденная гемофилия встречается почти у 70 % пациентов. В этом случае наследуется форма и тяжесть гемофилии. Около 30% наблюдений приходится на спорадические формы гемофилии, связанные с мутацией в локусе, кодирующем синтез плазменных факторов свертывания крови на Х-хромосоме. В дальнейшем такая спонтанная форма гемофилии становится наследственной.
Свертываемость крови, или гемостаз, служит важнейшей защитной реакцией организма. Активизация системы гемостаза происходит в случае повреждения сосудов и начала кровотечения. Свертываемость крови обеспечивается тромбоцитами и особыми веществами – плазменными факторами. При дефиците того или иного фактора свертывания своевременный и адекватный гемостаз становится невозможным. При гемофилии в связи с дефицитом VIII, IX или других факторов нарушается первая фаза свертывания крови - образование тромбопластина. При этом увеличивается время свертывания крови; иногда кровотечение не останавливается в течение нескольких часов.
Классификация гемофилии
В зависимости от дефицита того или иного фактора свертываемости крови, различают гемофилию А (классическую), В (болезнь Кристмаса), С и др.
- Классическая гемофилия составляет подавляющее большинство (около 85%) случаев синдрома и связана с дефицитом VIII фактора свертывания (антигемофильного глобулина), приводящим к нарушению образование активной тромбокиназы.
- При гемофилии В, составляющей 13% случаев заболевания, имеет место недостаток IX фактора (плазменного компонента тромбопластина, фактора Кристмаса), также участвующего в образовании активной тромбокиназы в I фазе свертывания крови.
- Гемофилия С встречается с частотой 1-2% и обусловлена недостаточностью XI фактора свертывания крови (предшественника тромбопластина). На остальные разновидности гемофилии приходится менее 0,5% случаев; при этом может отмечаться дефицит различных плазменных факторов: V (парагемофилия), VII (гипопроконвертинемия), Х (болезнь Стюарта – Прауэр) и др.
Тяжесть клинического течения гемофилии зависит от степени недостаточности коагуляционной активности плазменных факторов свертывания крови.
- При гемофилии тяжелой степени уровень недостающего фактора составляет до 1%, что сопровождается развитием тяжелого геморрагического синдрома уже в раннем детском возрасте. У ребенка с тяжелой гемофилией возникают частые спонтанные и посттравматические кровоизлияния в мышцы, суставы, внутренние органы. Сразу после рождения ребенка могут обнаруживаться кефалогематомы, длительные кровотечения из пуповинного отростка, мелена; позднее - продолжительные кровотечения, связанные с прорезыванием и сменой молочных зубов.
- При среднетяжелой степени гемофилии у ребенка уровень плазменного фактора составляет 1-5%. Заболевание развивается в дошкольном возрасте; геморрагический синдром выражен умеренно, отмечаются кровоизлияния в мышцы и суставы, гематурия. Обострения случаются 2-3 раза в год.
- Легкая форма гемофилии характеризуется уровнем фактора выше 5%. Дебют заболевания возникает в школьном возрасте, часто в связи с травмами или операциями. Кровотечения более редкие и менее интенсивные.
Симптомы гемофилии
У новорожденных детей признаками гемофилии могут служить длительное кровотечение из культи пуповины, подкожные гематомы, кефалогематомы. Кровотечения у детей первого года жизни могут быть связаны с прорезыванием зубов, оперативными вмешательствами (инцизией уздечки языка, циркумцизио). Острые края молочных зубов могут стать причиной прикусывания языка, губ, щек и кровотечений из слизистых оболочек полости рта. Однако в грудном возрасте гемофилия дебютирует редко в связи с тем, что материнском молоке содержится достаточное количество активной тромбокиназы.
Вероятность посттравматических кровотечений значительно возрастает, когда ребенок с гемофилией начинает вставать и ходить. Для детей после года характерны носовые кровотечения, подкожные и межмышечные гематомы, кровоизлияния в крупные суставы. Обострения геморрагического диатеза случаются после перенесенных инфекций (ОРВИ, ветрянки, краснухи, кори, гриппа и др.) вследствие нарушения проницаемости сосудов. В этом случае нередко возникают самопроизвольные диапедезные геморрагии. Ввиду постоянных и длительных кровотечений у детей с гемофилией имеется анемия различной степени выраженности.
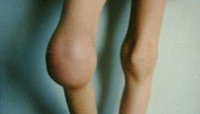
По степени убывания частоты кровоизлияния при гемофилии распределяются следующим образом: гемартрозы (70—80%), гематомы (10-20%), гематурия (14-20%), желудочно-кишечные кровотечения (8%), кровоизлияния в ЦНС (5%).
Гемартрозы являются наиболее частым и специфическим проявлением гемофилии. Первые внутрисуставные кровоизлияния у детей с гемофилией случаются в возрасте 1-8 лет после ушибов, травм или спонтанно. При гемартрозе выражен болевой синдром, отмечается увеличение сустава в объеме, гиперемия и гипертермия кожи над ним. Рецидивирующие гемартрозы приводят к развитию хронического синовита, деформирующего остеоартроза и контрактур. Деформирующий остеоартроз приводит к нарушению динамики опорно-двигательного аппарата в целом (искривлению позвоночника и таза, гипотрофии мышц, остеопорозу, вальгусной деформации стопы и др.) и к наступлению инвалидности уже в детском возрасте.
При гемофилии часто возникают кровоизлияния в мягкие ткани – подкожную клетчатку и мышцы. У детей обнаруживаются непроходящие синяки на туловище и конечностях, часто возникают глубокие межмышечные гематомы. Такие гематомы склонны к распространению, поскольку излившаяся кровь не сворачивается и, проникая вдоль фасций, инфильтрирует ткани. Обширные и напряженные гематомы могут сдавливать крупные артерии и периферические нервные стволы, вызывая интенсивные боли, паралич, атрофию мышц или гангрену.
Довольно часто при гемофилии возникают кровотечения из десен, носа, почек, органов ЖКТ. Кровотечение может быть инициировано любыми медицинскими манипуляциями (внутримышечной инъекцией, экстракцией зуба, тонзиллэктомией и др.). Крайне опасными для ребенка с гемофилией являются кровотечения из зева и носоглотки, поскольку могут привести к обструкции дыхательных путей и потребовать экстренной трахеостомии. Кровоизлияния в мозговые оболочки и головной мозг приводят к тяжелым поражениям ЦНС или летальному исходу.
Гематурия при гемофилии может возникать самопроизвольно или вследствие травм поясничной области. При этом отмечаются дизурические явления, при образовании кровяных сгустков в мочевыводящих путях - приступы почечной колики. У больных с гемофилией нередко обнаруживаются пиелоэктазия, гидронефроз, пиелонефрит.
Желудочно-кишечные кровотечения у пациентов с гемофилией могут быть связаны с приемом НПВС и др. лекарств, с обострением латентного течения язвенной болезни желудка и двенадцатиперстной кишки, эрозивным гастритом, геморроем. При кровоизлияниях в брыжейку и сальник развивается картина острого живота, требующая дифференциальной диагностики с острым аппендицитом, кишечной непроходимостью и др.
Характерным признаком гемофилии является отсроченный характер кровотечения, которое обычно развивается не сразу после травмы, а через некоторое время, иногда спустя 6-12 и более часов.
Диагностика гемофилии
Диагностика гемофилии проводится при участии ряда специалистов: неонатолога, педиатра, генетика, гематолога. При наличии у ребенка сопутствующей патологии или осложнений основного заболевания проводятся консультации детского гастроэнтеролога, детского травматолога-ортопеда, детского отоларинголога, детского невролога и др.
Супружеские пары, находящиеся в группе риска по рождению ребенка с гемофилией, должны пройти медико-генетическое консультирование еще на этапе планирования беременности. Выявить носительство дефектного гена позволяет анализ генеалогических данных и молекулярно-генетическое исследование. Возможно проведение пренатальной диагностики гемофилии с помощью биопсии хориона или амниоцентеза и исследования ДНК клеточного материала.
После рождения ребенка диагноз гемофилии подтверждается с помощью лабораторных исследований гемостаза. Основные изменения показателей коагулограммы при гемофилии представлены увеличением времени свертывания крови, АЧТВ, тромбинового времени, МНО, времени рекальцификации; уменьшением ПТИ и др. Решающее значение при диагностике формы гемофилии принадлежит определению снижения прокоагулянтной активности одного из факторов свертывания ниже 50%.
При гемартрозах ребенку с гемофилией проводится рентгенография суставов; при внутренних кровотечениях и забрюшинных гематомах – УЗИ брюшной полости и забрюшинного пространства; при гематурии – общий анализ мочи и УЗИ почек и т. д.
Лечение гемофилии
При гемофилии полное избавление от заболевания невозможно, поэтому основу лечения составляет заместительная гемостатическая терапия концентратами VIII и IX факторов свертывания крови. Необходимая доза концентрата определяется степенью выраженности гемофилии, тяжестью и видом кровотечения.
При незначительных наружных кровотечениях (порезах, кровотечениях из полости носа и рта) может использоваться гемостатическая губка, наложение давящей повязки, обработка раны тромбином. При неосложненном кровоизлиянии ребенку необходим полный покой, холод, иммобилизация больного сустава гипсовой лонгетой, в дальнейшем – УВЧ, электрофорез, ЛФК, легкий массаж. Больным с гемофилией рекомендуется диета, обогащенная витаминами А, В, С, D, солями кальция и фосфора.
Прогноз и профилактика гемофилии
Длительная заместительная терапия приводит к изоиммунизации, образованию антител, блокирующих прокоагулянтную активность вводимых факторов, и неэффективности гемостатической терапии в обычных дозах. В таких случаях больному с гемофилией проводится плазмаферез, назначаются иммунодепрессанты. Поскольку больным с гемофилией проводится частое переливание компонентов крови, не исключается риск инфицирования ВИЧ-инфекцией, гепатитами В, С и D, герпесом, цитомегалией.
Легкая степень гемофилии не влияет на продолжительность жизни; при тяжелой гемофилии прогноз ухудшается при массивных кровотечениях, обусловленных операциями, травмами.
Профилактика предполагает проведение медико-генетического консультирования супружеских пар, имеющих отягощенный семейный анамнез по гемофилии. Дети, больные гемофилией, всегда должны иметь при себе специальный паспорт, где указан тип заболевания, группа крови и Rh-принадлежность. Им показан охранительный режим, профилактика травм; диспансерное наблюдение педиатра, гематолога, детского стоматолога и др. специалистов; наблюдение в условиях специализированного гемофильного центра.
Читайте также: