Наследственные заболевания крови реферат
Обновлено: 02.07.2024
Врожденные заболевания являются одной из распространенных причин младенческой и детской смертности, развития хронических болезней у детей и инвалидизации.
Быстрый переход
Врожденные заболевания могут быть обусловлены генетическими мутациями (передающимися по наследству или спонтанными), инфекциями матери во время беременности (цитомегаловирус, ветряная оспа, краснуха), воздействием лекарств и химических веществ, загрязняющих воздух, воду или пищу. Причины множества врожденных дефектов до сих пор неизвестны.
Генетическими или наследственными факторами обусловлены около 20 % врожденных заболеваний. К ним относятся нарушения, при которых мутация затрагивает один ген (серповидно-клеточная анемия); хромосомные нарушения, при которых хромосомы (или их части) отсутствуют (синдром Тернера) или имеют структурные изменения (увеличение количества хромосом или трисомия при синдроме Дауна); многофакторные нарушения, вызванные мутациями двух и более генов. Врожденные дефекты и нарушения развития могут вызывать делеции или дупликации отдельных генов (изменения митохондриальной ДНК). Примером такого заболевания является муковисцидоз, характеризующееся поражением экзокринных желез и жизненно-важных органов (легких и желудочно-кишечного тракта). Врожденные заболевания также ассоциированы со случайными (новыми) повреждениями генов, спонтанными (не наследующимися от родителей) мутациями (большинство случаев ахондроплазии).
По статистике, врожденные заболевания имеют 2–3 % младенцев. К возрасту 1 года их число увеличивается до 5 %, поскольку не все эти патологии диагностируются сразу после рождения.
Врожденные (наследственные) заболевания также классифицируют по типу наследования. При аутосомно-доминантном наследовании заболевание может передаваться от родителя к ребенку в 50 % случаев (мышечная дистрофия Дюшенна, хорея Гентингтона). При аутосомно-рецессивном наследовании генетическая аномалия передается ребенку только в том случае, если оба родителя наделены одним и тем же дефектным геном (муковисцидоз, серповидно-клеточная анемия). Здесь частота наследования составит 25 %, то есть в среднем у 1 ребенка из 4 детей этих родителей будет аутосомно-рецессивное заболевание.
Патологические состояния могут передаваться и при наследовании, сцепленном с полом (наследование гена, находящегося в половых хромосомах). Х-сцепленные рецессивные заболевания почти всегда ограничены мужским полом (гемофилия, дальтонизм, мышечная дистрофия Дюшенна). Х-сцепленные доминантные заболевания встречаются как у мальчиков, так и у девочек, однако у детей мужского пола имеют более тяжелое течение (Х-сцепленный гипофосфатемический рахит). Y-сцепленные заболевания встречаются довольно редко, поскольку Y-хромосома содержит всего несколько генов (ихтиоз).
Некоторые врожденные заболевания формально не относятся к генетическим, но имеют ту или иную выраженность наследования: наследуются факторы риска либо сам ген, но с низкой пенетрантностью (частотой проявления гена в признаках).
Гемофилия
Гемофилия — группа редких наследственных нарушений свертываемости крови, вызванных дефицитом необходимого белка (фактора свертывания крови).
Гемофилия A (классическая) встречается чаще (>80 % случаев) и связана с дефицитом VIII фактора свертывания крови, гемофилия B (болезнь Кристмаса) — реже (>10 % случаев), она обусловлена недостаточностью IX фактора свертывания крови. Гемофилия C встречается очень редко, обусловлена дефицитом XI фактора свертывания крови, чаще всего в классификации группы ее не упоминают.
Заболевание относится к X-сцепленным с полом, наследуется по рецессивному признаку по женской линии. Классическую гемофилию вызывают мутации гена F8, расположенного на X-хромосоме. Примерно в 70 % случаев заболевание наследуется по Х-сцепленному образцу, в остальных случаях оно возникает спонтанно (новая мутация), в дальнейшем спонтанное заболевание становится наследственным. Гемофилией A болеют практически исключительно мужчины, редкие случаи заболевания у женщин, носительниц дефектного гена, почти всегда характеризуются легким течением. Гемофилию B вызывают мутации гена F9, так же расположенного на X-хромосоме, она характерна для мужчин, женщины болеют очень редко. В некоторых случаях заболевание возникает спонтанно (приобретенная гемофилия A или B) и тоже связано с недостаточностью VIII или IX факторов свертывания крови. Приобретенная гемофилия является аутоиммунным заболеванием, при котором организм вырабатывает антитела, атакующие факторы свертывания крови (чаще всего VIII фактор). Примерно в половине случаев приобретенной гемофилии у пациента имеется связанное с ней основное состояние или заболевание (беременность, аутоиммунные заболевания, миелопролиферативные заболевания, воспалительные заболевания кишечника и др.), в остальных эпизодах причина остается невыясненной.
Гемофилия A затрагивает примерно 1 из 5 000 новорожденных мальчиков, гемофилия B — примерно 1 из 25 000. Около 60 % пациентов имеют тяжелую форму гемофилии, обычно им ставят диагноз при рождении или в течение первых 2 лет жизни.
Возраст манифестации гемофилии и тяжесть течения заболевания зависят от уровня активности факторов свертывания крови. Легкая форма характеризуется уровнем фактора свертывания крови (VIII или IX), превышающим 5 % от нормы, средняя — 1–5 %, тяжелая — ниже 1 %. У большинства пациентов, независимо от тяжести течения заболевания, эпизоды кровотечения чаще встречаются в раннем детском, детском и подростковом возрасте, чем в дальнейшей взрослой жизни.
При легкой и умеренной формах гемофилии длительные кровотечения могут возникнуть только в результате травмы, хирургического вмешательства или стоматологической процедуры. Нередко диагноз гемофилии ребенку устанавливают к 5–6 годам, обратив внимание на длительное посттравматическое кровотечение, длительное кровотечение во время стоматологического лечения или операции. К другим заметным симптомам легкого и умеренного течения гемофилии относятся непроходящие гематомы (синяки), частые носовые кровотечения, кровоточивость десен.
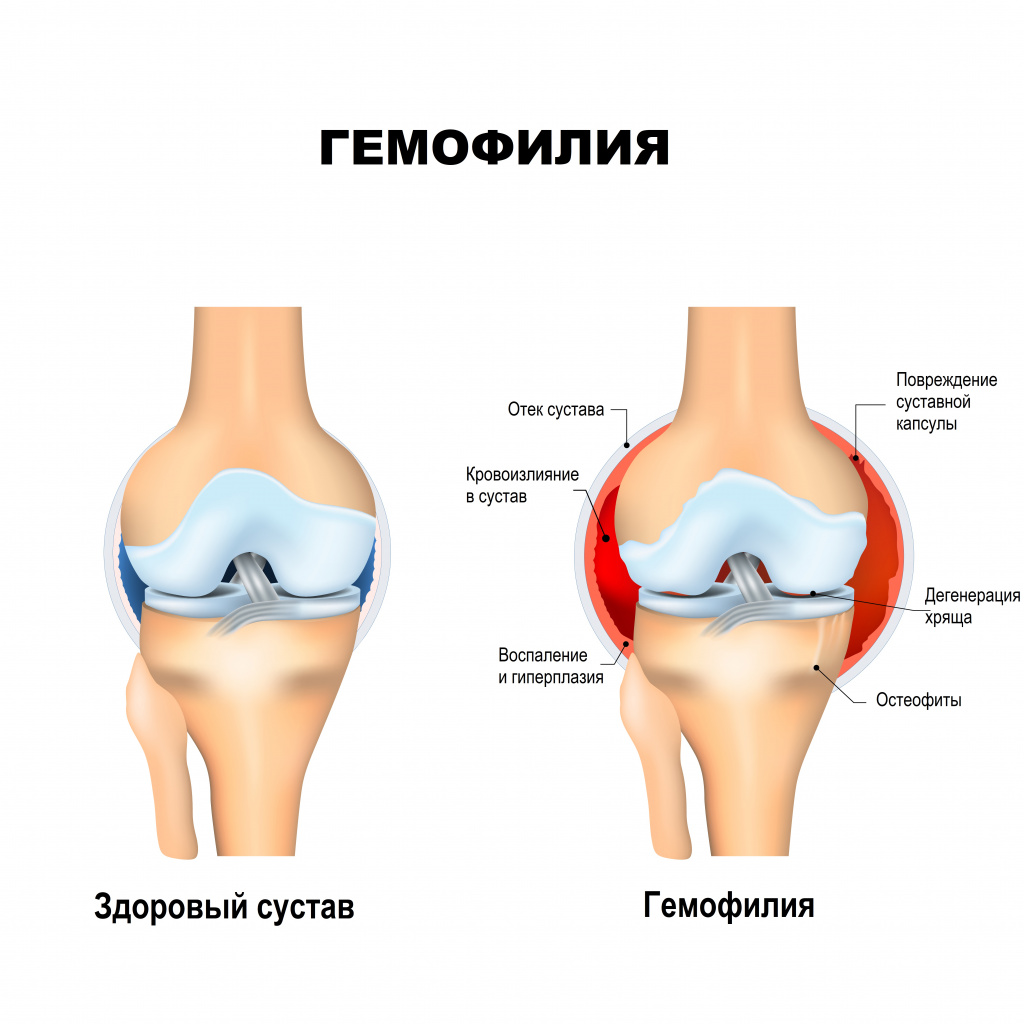
Для тяжелой формы гемофилии характерны эпизоды спонтанного кровотечения, которые приводят к кровоизлияниям различной локализации — в мягкие ткани, мышцы, суставы. При гемартрозах (кровоизлияние в полость сустава) возникает ограничение подвижности суставов, сопровождающееся острой болью и воспалением. У пациентов с тяжелой формой гемофилии спонтанные кровотечения чаще всего происходят в мышцы и суставы, однако могут затрагивать любой внутренний орган, включая почки, органы ЖКТ, головной мозг (гематурия, мелена, гематохезия, желудочно-кишечные кровотечения, внутричерепные кровотечения). Тяжелая форма гемофилии A обычно проявляется в раннем детском возрасте, диагноз чаще всего устанавливается к 2 годам ребенка. Обычными симптомами (при отсутствии лечения заболевания) здесь являются кровотечения из-за незначительных травм ротовой полости (прикусывание губ, языка, щек), подкожные гематомы, большие шишки после удара головой, спонтанные кровотечения (2–5 эпизодов в месяц).
Диагноз гемофилии A или B устанавливается на основании симптомов, истории личного и семейного анамнеза пациента, лабораторных исследований (общего анализа крови, коагулограммы, оценивающей состояние системы свертывания крови и активности ее белков, молекулярно-генетического исследования). Парам из группы риска по рождению ребенка с гемофилией необходимо получить генетическую консультацию на этапе планирования беременности. Определение конкретной мутации гена F8 (или F9) помогает не только выявить женщин-носительниц дефектного гена в конкретной семье, но и полезно для пренатальной диагностики гемофилии (амниоцентез, биопсия хориона).
Некоторым пациентам с легкой формой гемофилии назначается синтетический аналог вазопрессина — десмопрессин, повышающий VIII фактор свертывания крови (вводится внутривенно или интраназально), а также антифибринолитические средства, замедляющие распад факторов свертывания крови.
Примерно в 30 % случаев лечения тяжелых случаев гемофилии длительная заместитетельная терапия может приводить к изоиммунизации — образованию антител к вводимым факторам свертывания крови (иммунная система распознает вводимый фактор VIII как чужеродный). Этот процесс может сопровождаться аллергическими реакциями (разной степени тяжести), возрастает риск жизнеугрожающих кровотечений. В этом случае пациенту назначается альтернативное лечение — плазмаферез, иммунодепрессанты.
Гемохроматозы
Гемохроматоз — наследственное заболевание, характеризующееся нарушением обмена железа и его накоплением в тканях и органах.
Заболевание сопровождается повышенным всасыванием железа в желудочно-кишечном тракте и накоплением в печени, сердце, поджелудочной железе, суставах, гипофизе, что приводит к полиорганной недостаточности и развитию таких болезней, как цирроз печени, рак печени, диабет, болезни сердца и суставов.
Наследственный гемохроматоз вызывают мутации генов HFE, HFE2, HAMP, SLC40A1 и TfR2, он классифицируется в зависимости от возраста начала, генетической причины и способа наследования: гемохроматоз 1 типа, гемохроматоз 2 типа, гемохроматоз 3 типа, гемохроматоз 4 типа. Вторичный гемохроматоз ассоциирован с другими заболеваниями (не является наследственным) — анемией, хронической болезнью печени, инфекциями.
Гемохроматоз, редко возникающий в младенческом возрасте и не имеющий явной причины, называют неонатальным гемохроматозом. При этой форме заболевания избыточное накопление железа в тканях и органах начинается еще до рождения ребенка. Неонатальный гемохроматоз быстро прогрессирует и характеризуется поражениями печени, которые выявляют при рождении или в первые дни жизни. Дети с этим заболеванием часто рождаются недоношенными или имеют нарушения внутриутробного развития. Точная причина неонатального гемохроматоза неизвестна, есть версия, что он развивается в том случае, если иммунная система матери распознает клетки печени ребенка как чужеродные. Симптомы обычно включают гипогликемию, нарушения свертываемости крови, пожелтение кожных покровов и склер глаз, отеки. Диагноз устанавливается на основании признаков и симптомов, лабораторных и инструментальных исследований, биопсии печени. Лечение включает переливание крови, внутривенное введение иммуноглобулинов, трансплантацию печени.
Гемохроматоз 1 типа ассоциирован с мутациями в гене HFE, расположенном на коротком плече 6–й хромосомы, наследуется по аутосомно-рецессивному типу, является наиболее распространенным типом наследственного гемохроматоза и поражает в основном мужчин. Вероятность того, что ребенок унаследует мутацию в гене HFE от родителя составляет 25 %, вероятность того, что он станет носителем дефектного гена — 50 %.
Начальные симптомы заболевания обычно отмечаются в возрасте 40–60 лет и включают боль в животе, снижение полового влечения, усталость, слабость, боли в суставах, сухость кожи. В дальнейшем проявляются такие симптомы и осложнения, как изменение пигментации кожи (бронзовая кожа), выпадение волос на голове и туловище, аритмия, кардиомиопатия, хроническая сердечная недостаточность, сахарный диабет, гепатомегалия, цирроз печени, спленомегалия, атрофия яичек, аменорея (у женщин) и другие.
Диагноз устанавливается на основании симптомов заболевания, лабораторных исследований (уровень железа, ферритина, трансферрина, железосвязывающей способности сыворотки крови, маркеры функции печени, уровень глюкозы крови и др.), инструментальных исследований (рентгенография суставов, электрокардиография и эхокардиография сердца, УЗИ органов брюшной полости, МРТ печени и др.), биопсии печени, молекулярно-генетического исследования (в том числе для выявления родственников — имеющих гемохроматоз или носителей).
Лечение гемохроматоза направлено на удаление избытка железа из организма (флеботомия, хелатирование) с помощью забора крови (как при взятии анализов или донорстве крови, только в большем объеме) и специальных препаратов, образующих комплексное соединение с железом и способствующих его удалению из железосодержащих белков (дефероксамин).
Терапевтическая флеботомия сначала проводится 1–2 раза в неделю, поддерживающая — каждые 2–4 месяца. Вместе с этим проводится симптоматическое лечение сахарного диабета, патологий сердца, цирроза печени и других заболеваний, вызванных гемохроматозом. Пациентам запрещен прием препаратов железа и любых медикаментозных средств или комплексов, которые могут содержать железо и витамин C, запрещен алкоголь во избежание дальнейшего повреждения печени (если оно имеется), рекомендуется придерживаться диеты, исключающей продукты с высоким содержанием железа (красное мясо, яблоки, печень, шпинат).
Гемохроматоз 2 типа вызывают мутации в генах HFE2 или HAMP, он наследуется по аутосомно-рецессивному типу и проявляется чаще всего в детском возрасте. Для этого типа заболевания характерны следующие симптомы: высокие уровни ферритина и трансферрина, врожденный фиброз печени, артропатия, кардиомиопатия, генерализованная гиперпигментация кожи, мышечная слабость, гипогонадизм, задержка полового созревания, сахарный диабет, цирроз печени, остеопороз, спленомегалия, гепатомегалия и другие. Диагностика и лечение гемохроматоза 2 типа проводятся аналогично диагностике и лечению гемохроматоза 1 типа.
Гемохроматоз 3 типа вызывают мутации в гене TFR2, он наследуется по аутосомно-рецессивному типу. Симптомы обычно проявляются до 30 лет и, помимо вышеперечисленных, могут включать снижение уровня лимфоцитов и нейтрофилов в крови, красные или пурпурные пятна на коже. Диагностика и лечение гемохроматоза 3 типа проводятся аналогично диагностике и лечению гемохроматоза 1 и 2 типов.
Симптомы гемохроматоза 4 типа могут проявиться как в детском, так и во взрослом возрасте. Этот тип заболевания вызывают мутации гена SLC40A1, наследуется оно по аутосомно-доминатному типу. Вероятность того, что ребенок унаследует мутацию в гене SLC40A1 от родителя составляет 50 %, соответственно, вероятность того, что он станет носителем дефектного гена — тоже 50 %. Симптомы гемохроматоза 4 типа могут проявляться в виде повышенной утомляемости, слабости, болей в суставах, изменения пигментации кожи, затруднения дыхания, сердечной недостаточности, сахарного диабета, анемии, врожденного фиброза печени, цирроза печени, остеоартроза, катаракты. Диагностика и лечение гемохроматоза 4 типа проводятся аналогично диагностике и лечению гемохроматоза 1, 2 и 3 типов.
Наследственный гемохроматоз — заболевание с пониженной пенетрантностью (вероятностью того, что ген будет иметь любые фенотипические проявления) дефектного гена, т. е. у некоторых людей с мутациями гена HFE никогда не проявятся симптомы, при этом у их детей или других членов семьи с мутацией гена произойдет манифестация заболевания.
Мышечная дистрофия Дюшенна
Мышечная дистрофия Дюшенна — редкое наследственное заболевание, характеризующееся нарастающей мышечной слабостью с последующей атрофией мышц. Ассоциировано с мутацией гена DMD, расположенного на половой X-хромосоме (X-сцепленный рецессивный тип наследования).
Система свертывания крови человека представляет собой многокомпонентный и чрезвычайно сложный механизм, играющий важнейшую роль в защите всего организма.
Указанный механизм представлен тремя звеньями: сосудистым, тромбоцитарным и плазменно-коагуляционным. Заболевания, связанные с неспособностью факторов свертывания крови обеспечивать нормальный процесс образования сгустка, составляют обширную группу коагулопатий. Эта группа болезней представлена наследственными коагулопатиями и множеством приобретенных форм коагулопатий, которые являются следствием других заболеваний: цирроз и рак печени, аутоиммунные (системная красная волчанка, иммунная тромбоцитопеническая пурпура, геморрагический васкулит и т.п.), инфекционные заболевания, токсическое воздействие лекарственных средств и ядов, наследственные заболевания обмена веществ.
Наследственные заболевания гемостаза
Гемофилия и болезнь Виллебранда являются наиболее частыми наследственными заболеваниями системы плазменного звена гемостаза. Фактор Виллебранда и фактор VIII в плазме крови представлены в виде молекулярного комплекса, при этом фактор Виллебранда выполняет защитную роль для фактора VIII, оберегая его от разрушения протеином С.
Именно поэтому при отсутствии фактора Виллебранда уровень фактора VIII может быть значительно снижен. Таким образом, несмотря на различие данных заболеваний, они проявляют определенный молекулярно-биологический синергизм. В этой связи для практикующих врачей целесообразно рассмотреть два заболевания, занимающих более 90% наследственных коагулопатий, обусловленных дефицитом или функциональной несостоятельностью пламенных факторов свертывания крови — VIII, IX (гемофилия А или В соответственно), или фактора Виллебранда (болезнь Виллебранда).
Крайне редко встречается и дефицит фактора VII. Гипопроконвертинемия проявляется геморрагическим синдромом в зависимости от выраженности дефицита фактора VII. В тяжелых случаях отмечаются гемартрозы, гематомы, у женщин меноррагии.
И встречаются, и диагностируются нечасто
В целом наследственные коагулопатии встречаются нечасто, а диагностируются еще реже. На это есть две основные причины: низкая распространенность, часто сопряженная с субклиническим течением заболевания (при болезни Виллебранда) и недостаточно развитая система лабораторной диагностики нарушений гемостаза в большинстве регионов нашей страны. Количество больных гемофилией в России составляет чуть более 7,5 тыс. человек, а с болезнью Виллебранда должно быть около 16 тыс. Точная цифра не установлена. Редкие наследственные формы коагулопатий — гипопроконвертинемия (дефицит фактора VII), гипо- и афибриногенемия, дефицит факторов XII, XIII, XI, V встречается крайне редко.
Наследственные коагулопатии неизлечимы, но возможности современной препаратной терапии позволяют обеспечить больным продолжительность и качество жизни, сравнимые с таковой в общей популяции.
Эти гены локализованы на длинном плече X-хромосомы и наследуются по рецессивному признаку, передаваясь через женщин только детям мужского пола. В популяции уровень факторов VIII и IX варьирует от 100 ± 50%, но у женщин — кондукторов гемофилии— он может быть ниже нормы, вслед-ствие функциональной неполноценности одного из двух генов. Тяжесть гемофилии зависит от уровня активности фактора VIII или IX. При тяжелой форме гемофилии фактор VIII (или IX) отсутствует или проявляет остаточную активность (менее 2%). В этом случае заболевание обычно проявляется с раннего детства.
Характерны кровотечения при нарушении целостности слизистых и кожных покровов, гематомы (кровотечение из пуповины, кефалогематомы, экхимозы). Когда ребенок начинает ходить, появляются первые кровоизлияния в суставы. При средней форме (от 2 до 5%) также отмечается поражение опорно-двигательной системы, а при легкой (более 5%) заболевание обычно проявляется при травмах и хирургических операциях, которые могут сопровождаться сильными кровотечениями из-за быстрого истощения эндогенного фактора VIII или IX. Поздняя диагностика заболевания может привести к трагическим последствиям.
Сегодня такого сценария можно избежать
Возникновение первичного гемартроза даже после его видимого устранения вызывает невидимые изменения в хрящевой ткани сустава, которые можно зарегистрировать на ЯМР-томограмме. Распавшиеся эритроциты формируют среду для возникновения вторичного асептического воспалительного процесса, а образовавшийся гемосидерин откладывается в хрящевой ткани суставных поверхностей.
Известно, что полупериод инактивации фактора VIII и IX (T?) невелик. В среднем он составляет соответственно 12 и 24 часа, однако у каждого пациента он может значительно варьировать. При гемофилии А такой разброс для фактора VIII составляет от 7 до 20 часов. Этот показатель важно учитывать при коррекции системы гемостаза у больных гемофилией, особенно, если лечение проводится в течение длительного времени.
Самое уязвимое звено в лечении этих пациентов — определение потребности при назначении факторов свертывания крови. Все существующие факторы свертывания крови вводятся внутривенно, т.е. их биодоступность составляет 100%, однако катаболизм этих сложных белковых структур зависит от многих индивидуальных параметров всей системы гемостаза.
На сегодняшний день профилактическая заместительная терапия пациентам с наслед-ственными коагулопатиями подбирается эмпирически, чаще руководствуясь только первоначально поставленным диагнозом и видимым клиническим результатом, который оценивается весьма субъективно. То же самое касается и однократных лечебных доз, направленных на купирование геморрагического эпизода. Для широкой клинической практики подобные тесты представляются затруднительными, во всяком случае, на сегодняшний день.
Лечение по требованию и по ситуации
Для того, чтобы формализировать стандарты терапии больных гемофилией, в 2005 году был разработан и утвержден Протокол ведения больных гемофилией. Это событие принципиально изменило качество жизни пациентов.
Лечение заболевания может осуществляться не только с целью профилактики кровоизлияний. Протокол ведения больных c гемофилией предусматривает модель лечения состоявшихся кровотечений или кровоизлияний. В этом случае пиковый уровень фактора VIII или IX через 30 минут после инъекции должен составлять 40—100% в зависимости от клинической ситуации.
Инъекции препарата повторяются каждые 12 часов при гемофилии А и каждые 24 часа при гемофилии В в дозе ? от первоначальной до исчезновения симптомов кровоизлияния. Как показывает наш опыт, такая схема лечения приемлема для пациентов с единичными геморрагическими осложнениями. Как и при многих других заболеваниях, при гемофилии легче профилактировать кровоизлияния, а не лечить их осложнения.
Документ общерекомендательного характера
Тем не менее лечащим врачам-гематологам надлежит помнить, что данный документ носит общерекомендательный характер, а подход к лечению каждого пациента должен быть индивидуальным, учитывая порой психоэмоциональный профиль пациента и даже членов его семьи.
Наследственные коагулопатии неизлечимы, но возможности современной препаратной терапии позволяют обеспечить больным продолжительность и качество жизни, сравнимые с таковой в общей популяции.
Внутривенное введение препарата осуществляется самим пациентом или его род-ственниками после прохождения специализированной программы обучения, а наблюдение за пациентом осуществляет центр гемофилии или врач-гематолог.
Установлено также, что фенотип заболевания может отличаться от его генотипа. Не все больные с одинаковым уровнем фактора VIII или IX требуют одинакового лечения. 10—15% больных с тяжелой формой гемофилии А вообще не нуждаются в профилактическом лечении, т.е. в постоянном введении препаратов, однако все без исключения больные гемофилией нуждаются в пожизненном обеспечении факторами свертывания крови VIII или IX. Это касается и других наслед-ственных форм коагулопатий — болезни Виллебранда и гипопроконвертинемии (дефицит фактора VII).
Современные антигемофильные препараты факторов свертывания крови VIII и IX, с точки зрения этиотропной терапии, направленной на достижение клинического результата, не обладают между собой значительными различиями. Все они компенсируют уровень недостающего фактора свертывания крови в равной степени. Их активность выражается в стандартных международных единицах (МЕ). Условно они могут классифицироваться на факторы свертывания крови VIII содержащие фактор Виллебранда и не содержащие. Количество и качество фактора Виллебранда может значительно отличаться в них. Существуют препараты, в которых содержание фактора Виллебранда повышено. Есть изолированный фактор Виллебранда, не содержащий фактор VIII.
Рекомбинантные факторы свертывания крови
К препаратам плазмы фактора VIII, не содержащим фактора Виллебранда, количество которого у больных гемофилией не изменено, относятся препараты, прошедшие аффинную хроматографию, а также генно-инженерные (рекомбинантные) факторы свертывания крови VIII (МНН: октоког альфа). Группа препаратов октоког альфа представлена препаратами трех поколений (классификация условна): первое, содержащее добавленный альбумин, необходимый для стабилизации молекулы фактора VIII, второе — содержащее следы человеческого альбумина, и третье — свободное от присутствия альбумина.
Основой эффективной терапии наследственных коагулопатий является: ранняя диагностика, выбор правильной модели лечения и полноценное, беспрерывное обеспечение больных факторами свертывания крови VIII или IX. Если количество препарата недостаточно для лечения, заболевание начинает неуклонно прогрессировать и может свести на нет все ранее предпринятые усилия.
Существует рекомбинантный фактор VIII с модифицированной молекулой, т.е. удаленным гликопротеидным фрагментом (мороктоког альфа). Опыт его использования не так объемен, как у предыдущих препаратов. Для больных с гемофилией В могут быть использованы препараты фактора свертывания крови IX — рекомбинантные (наноког альфа) и полученные из донорской плазмы, а также препараты протромбинового комплекса (PPSB).
У больных с болезнью Виллебранда более целесообразно применять препараты, содержащие физиологическое или больше физиологического соотношения фактора Виллебранда к фактору VIII. Эти препараты были разработаны специально для лечения больных с болезнью Виллебранда, и содержание последнего указывается на этикетке флакона.
Почти абсолютная вирусная безопасность
Осложнения при гемофилии, как след-ствие лечения, также неизбежны. Наиболее опасное, но редко встречающееся в России (около 3—5% больных гемофилией А) — появление резистентности к терапии, обусловленное образованием иммуноглобулинов, чаще G класса, т.н. аутоантител, которые избирательно блокируют прокоагулянтную активность молекулы фактора VIII. При гемофилии В образование антител отмечается крайне редко (менее 1—2%).
Описаны случаи образования антител и к фактору Виллебранда. К фактору VII антитела практически не образуются. Для практикующих гематологов важно знать, что антигенная стимуляция (введение препарата) может стимулировать иммунный ответ и вызвать полную толерантность к заместительной терапии. Заподозрить ингибитор стоит в случае неэффективности эффективной ранее терапии. Этиология и патогенез этого явления остаются неясными.
Диагностика в регионах недоступна
При болезни Виллебранда, характеризующейся снижением активности фактора Виллебранда, ристоцетин-индуцированной агрегации тромбоцитов, часто со снижением уровня фактора VIII и антигена фактора VIII, важно определить следующее: имеет ли место недостаток выработки фактора Виллебранда (количественный дефицит — тип I) или синтезируемая структура молекулы фактора неполноценна (качественный дефицит — тип II).
Диагностика заболевания достаточно сложна и часто недоступна в регионах, т.к. требует проведения сложных коагулологических исследований. Важными симптомами заболевания являются длительные носовые кровотечения, у женщин затяжные mensis, которые могут приводить к железодефицитной анемии, длительные кровотечения после удаления зуба или незначительных хирургических вмешательств и проявление таких же симптомов у близких родственников (отец, мать, брат, сестра). Пациенты с подобными симптомами должны быть обследованы в специализированных центрах.
Крайне редкое заболевание
Крайне редко встречающееся заболевание — наследственный дефицит фактора VII. При выраженном дефиците этого фактора отмечается тяжелый геморрагический синдром, сопровождающийся гематомами, гемартрозами, а у женщин — опасными для жизни меноррагиями.
Для лечения больных с гипопроконвертинемией может применяться фактор свертывания крови VII (плазменный) в дозе 30—40 МЕ/кг каждые 8—10 часов. При его отсутствии — эптаког альфа активированный в дозе 20—40 мкг/кг каждые 2—4 часа до полной остановки кровотечения.
Основой эффективной терапии наслед-ственных коагулопатий является: ранняя диагностика, выбор правильной модели лечения и полноценное, беспрерывное обеспечение больных факторами свертывания крови VIII или IX. Если количество препарата недостаточно для лечения, заболевание начинает неуклонно прогрессировать и может свести на нет все ранее предпринятые усилия.
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Гемофилия: причины появления, симптомы, диагностика и способы лечения.
Определение
Гемофилия является редким, но серьезным наследственным заболеванием, связанным с нарушением функции свертывания крови, что приводит к неконтролируемым и часто спонтанным кровотечениям и кровоизлияниям в различные органы и ткани. При гемофилии организм человека не может естественным способом остановить кровотечение, возникшее из-за повреждения кровеносного сосуда в результате травмы, хирургического вмешательства или стресса. Интенсивность кровотечения сопоставима с таковой у здорового человека, однако оно продолжается значительно дольше.
Кровь содержит много белков, называемых факторами свертывания, которые помогают остановить кровотечение. Существует два различных типа гемофилии. Каждый отличается дефицитом определенного фактора свертывания крови. Заболевание появляется в результате мутации гена фактора свертывания крови VIII (гемофилия А) или фактора свертывания IX (гемофилия В). Наиболее распространенным типом заболевания является гемофилия А, которая встречается у 80-85% больных гемофилией. В типичном случае человек с тяжелой формой гемофилии А страдает кровотечением 35 раз в год.
Тяжесть гемофилии определяется количеством фактора свертывания крови — чем он ниже, тем больше вероятность возникновения кровотечения, которое может привести к серьезным проблемам со здоровьем.
Гемофилия известна человечеству более 2000 лет. Первые сведения о заболевании содержатся в Талмуде, согласно которому мальчику не делали обрезание, если двое его старших братьев умерли из-за кровопотери, вызванной этой же манипуляцией.
Причины появления гемофилии
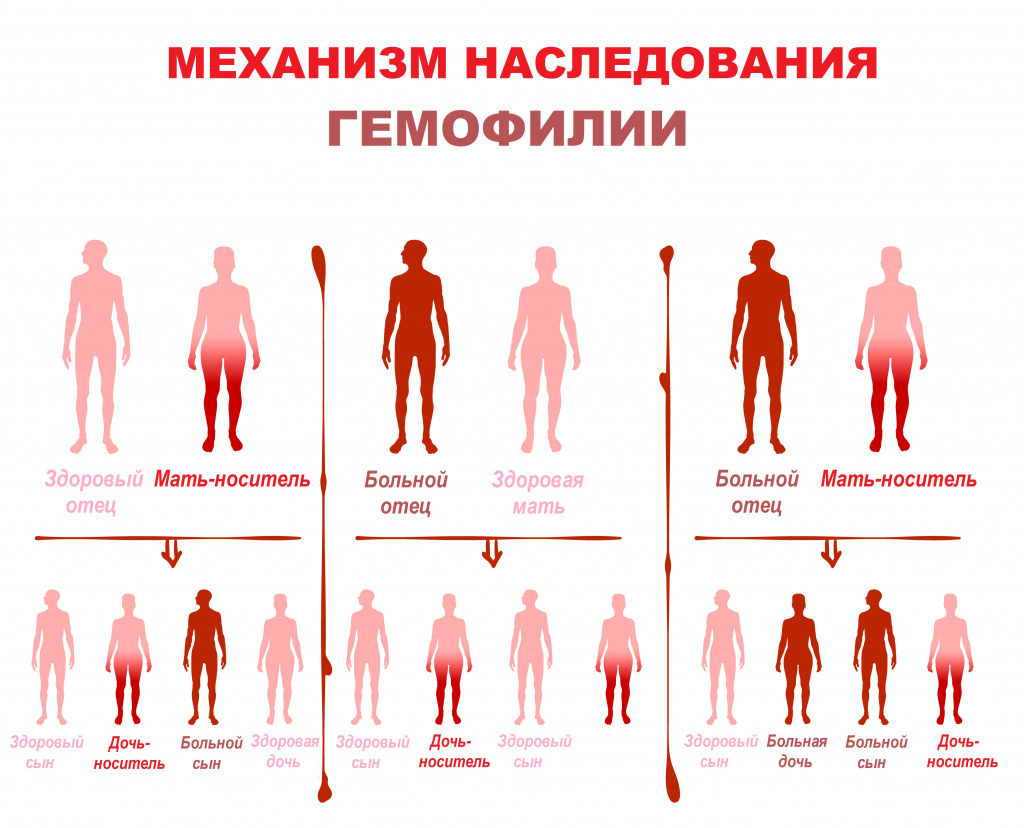
Заболевание передается от родителей к ребенку, хотя примерно в трети случаев вызвано спонтанной мутацией. Гены, кодирующие оба фактора свертывания крови, локализованы в Х-хромосоме. У женщин имеются две половые хромосомы ХХ. Хромосомный набор мужчин — ХY. При рождении девочек одна Х-хромосома наследуется от матери, а другая — от отца. Мужчина, больной гемофилией, имеет одну аномальную Х-хромосому и одну неповрежденную Y-хромосому. В случаях рождения мальчиков Y-хромосома наследуется от отца, а одна из Х-хромосом — от матери.
Сыновья мужчины, больного гемофилией, и здоровой женщины будут со 100% вероятностью здоровыми, а дочери — со 100% вероятностью носительницами гемофилии. В среднем, носительницы гемофилии будут иметь 30-50% от нормального уровня фактора свертывания.
Если женщина — носительница гемофилии имеет одну Х-хромосому здоровую, а другую аномальную, то от здорового мужчины у нее с равной долей вероятности могут родится как здоровые сыновья (50%), так и больные (50%) гемофилией, а дочери также с равной долей вероятности будут или здоровыми (50%), или носительницами (50%) гемофилии. Именно поэтому гемофилией страдают в основном пациенты мужского пола, которые наследуют пораженную X-хромосому от матери.
Дочери могут болеть гемофилией с вероятностью 25%, если она есть у отца, а мать – носительница гена. Но это случается крайне редко.
Классификация заболевания
Кодирование по Международной классификации болезней 10-го пересмотра:
- D66.0 – Наследственный дефицит фактора VIII.
- D67.0 – Наследственный дефицит фактора IХ.
- легкая – активность фактора 5-40% (0,05–0,40 МЕ/мл);
- средняя – активность фактора 1-5% (0,01–0,05 МЕ/мл);
- тяжелая – активность фактора менее 1% (менее 0,01 МЕ/мл).
- Типичные:
- гемартрозы крупных суставов 70-90%,
- гематомы (кровоизлияния в мышцы/мягкие ткани) 20-40%,
- кровотечения из слизистых (носовые, десневые, луночковые) 10%,
- гематурии 5-10%.
- в ЦНС – 5%,
- в ЖКТ – 5%,
- в области шеи/горла – 5%,
- забрюшинные гематомы – 5%.
Гемофилия средней тяжести имеет сходные проявления. Первые признаки, как правило, манифестируют после года. У пациентов с активностью факторов более 2% реже возникают кровоизлияния в суставы, забрюшинные гематомы и гематурии. Наиболее типичны посттравматические гематомы и длительные кровотечения, особенно при травмах слизистых оболочек.
Геморрагический синдром обычно возникает вследствие значительных травм или в результате хирургического лечения. Поражение опорно-двигательного аппарата встречается чрезвычайно редко.
Общие признаки гемофилии:
- длительные кровотечения после травм;
- внутрисуставные кровотечения, вызывающие отек и боль;
- кровоизлияние в кожу (гематомы) или мышцу и мягкие ткани;
- десневые кровотечения, которое трудно остановить после удаления или потери зуба;
- постинъекционные кровотечения;
- кровь в моче или кале;
- частые и трудно останавливаемые носовые кровотечения.
Диагностика гемофилии начинается с выявления наличия геморрагического синдрома в анамнезе у пациента и членов семьи. Сбор жалоб и анамнеза вкупе с физикальным осмотром позволяют определить необходимый объем обследования пациента.
Лабораторная диагностика
Рекомендуется проведение поэтапного лабораторного коагулологического исследования с целью верификации диагноза и исключения приобретенного дефицита фактора VIII или IX, а также исключения дефицита других факторов свертывания крови.
В качестве первого этапа выполняют коагулологический скрининг, в ходе которого определяются следующие показатели:
-
активированное частичное тромбопластиновое время;
АЧТВ – скрининговый тест для оценки внутреннего пути активации свертывания крови (факторы XII, XI, IX, VIII, X, V и II) и мониторинга пациентов, получающих гепариновую терапию.
Протромбин, МНО – один из важнейших показателей коагулограммы. Скрининговый тест для оценки внешнего пути активации свертывания (факторы II, VII, V и X).
Скрининговый тест для оценки эффективности заключительного этапа свертывания крови (образования фибрина из фибриногена).
Белок, предшественник фибрина, составляющего основу сгустка при свертывании крови.
Исследование применяют с целью диагностики полицитемий, тромбоцитопений, тромбоцитозов, для мониторинга терапии, для контроля уровня тромбоцитов при приеме ряда лекарственных препаратов.
-
активность факторов VIII, активность факторов IX;
Исследование применяют для диагностики гемофилии типа А, приобретенных гемофилических расстройств, при обследовании пациентов с удлиненным АЧТВ.
Исследование применяют для диагностики гемофилии типа B (болезни Кристмаса), приобретенных гемофилических расстройств, в обследованиях пациентов с удлиненным АЧТВ.
Показатель используют в диагностике и определении типа болезни Виллебранда, контроле лечения десмопрессином и заместительной терапии, а также в качестве одного из маркеров эндотелиальной дисфункции.
Исследование включает скрининговый и, при необходимости, подтверждающий (рефлексный) тесты с разведенным ядом гадюки Рассела (dRVTT - dilute Russell viper venom time).
Для исключения приобретенных дефицитов факторов VIII или IX требуется молекулярно-генетическая диагностика.
Генетическое исследование, направленное на поиск мутаций в гене фактора IX при гемофилии B – заболевании, характеризуемом гематомным типом кровоточивости.
Инструментальная диагностика позволяется визуализировать кровотечения/кровоизлияния различных локализаций, а также выявить осложнения, развившиеся в результате геморрагических проявлений. По показаниям проводятся следующие обследования:
Исследование слизистой оболочки верхнего отдела желудочно-кишечного тракта с возможностью выполнения биопсии или эндоскопического удаления небольших патологических образований.
Ультразвуковое сканирование структуры крупных суставов и определение их функциональной активности.
Сканирование внутренних органов брюшной полости для оценки его функционального состояния и наличия патологии.
Комплексное ультразвуковое сканирование органов мочевыделительной системы, позволяющее обнаружить патологию на ранних стадиях развития.
Безопасное и информативное сканирование структур головного мозга для диагностики его патологий.
Исследование, позволяющее получить данные о состоянии органов грудной клетки и средостения.
Сканирование головного мозга, черепа и окружающих их тканей, позволяющее диагностировать различные патологии.
Лечением больных гемофилией занимается врач-гематолог. Для подтверждения наличия геморрагических проявлений или их последствий также рекомендуется проведение консультации специалистов. По показаниям возможны консультации:
- неонатолога или врача-педиатра ;
- врача-генетика;
- травматолога-ортопеда;
- хирурга ;
- уролога ;
- невролога ;
- оториноларинголога ;
- стоматолога.
Гемофилия является неизлечимым заболеванием, поэтому основная цель терапии – купирование симптомов. Главный принцип лечения – специфическая заместительная терапия концентратами факторов свертывания. Концентраты фактора производятся либо из человеческой плазмы (плазматические), либо они генетически модифицированы (рекомбинантные).
Если препараты произведены из плазмы, принимается ряд мер, чтобы убедиться в том, что в продукте отсутствует вирусная инфекция, и что пациенты не заразятся такими вирусами, как гепатит С или ВИЧ. С этой целью применяются строгие критерии к выбору доноров плазмы. Кроме того, производители разработали различные методы по очищению и дезактивации вирусов во время производственного процесса препаратов крови. Тем не менее, пациентам следует знать, что есть теоретическая вероятность заражения через использование препаратов плазмы.
Рекомбинантные препараты создают из живых клеток, таких как моноклональные антитела. Они считаются технологически более продвинутыми и несут меньший риск вирусного заражения.
В настоящее время нет оснований для предпочтительного выбора между плазматическими или рекомбинантными препаратами.
Концентраты факторов свертывания крови вводятся внутривенно. Однако примерно у 30% пациентов с тяжелой гемофилией организм начинает вырабатывать антитела, что крайне затрудняет лечение, а в некоторых случаях делает его невозможным.
Существует два вида специфической терапии – профилактическая и лечение по факту возникновения кровотечений (по требованию). Во всех случаях рекомендовано сразу использовать достаточную дозу и соблюдать кратность введения препарата.
Для лечения гемофилии среди более новых лекарственных средств можно отметить фитусиран (снижает выработку природного антикоагулянтного белка – антитромбина) и концизумаб (увеличивает выработку тромбина). Генная терапия с использованием аденовирусного вектора для доставки гена фактора VIII или IX также подвергается клиническим испытаниям.
Серьезные осложнения встречаются при тяжелой и умеренной гемофилии и чаще всего проявляются в виде разрушений суставов и развития артрита. Кровотечения в сустав приводят к разрушению его нормальных тканей и развитию хронического артрита, очень болезненного и приводящего к нарушению функции сустава. Кровоизлияния в суставы отмечаются и у детей с 2-3 летнего возраста. Чаще всего поражаются крупные суставы – коленные, локтевые, голеностопные. Если кровотечение продолжается, то крайне высок риск инвалидизации и ограничения движения в суставе (в худшем случае человек может потерять конечность).
![Сустав.jpg]()
Помимо негативного влияния на качество жизни человека, кровоизлияния нередко становятся жизнеугрожающими, если происходят в жизненно важных органах, например, в мозге.
Профилактика гемофилии
Профилактическая заместительная терапия концентратами факторов свертывания крови – необходимое условие сохранения физического и психологического здоровья пациентов с тяжелой и среднетяжелой гемофилией. Профилактическое лечение обычно назначается детям, чтобы снизить риск кровотечений и повреждений суставов. В последнее время с этой же целью профилактику стали назначать и пожилым людям. Профилактика предполагает вливание фактора свертывания крови на регулярной основе (через день), чтобы сохранить нормальное свертывание крови у больного и предотвратить спонтанные кровотечения.
Детей с гемофилией необходимо оберегать от травм, предотвращать удаление зубов путем тщательной санации полости рта и квалифицированной стоматологической помощи.
Пациентам перед оперативным вмешательством или удалением зуба проводят заместительную терапию фактором (предпочтительно с использованием рекомбинантного препарата).
Молодые люди должны осознать, что поддержание физической активности очень важно для укрепления мышц, связок и суставов. Мышечная слабость, плохая координация движений значительно повышают вероятность травмы сустава и последующего развития воспаления (артрита). Хорошая физическая форма, крепкий мышечный каркас снижают риск спорадических кровотечений. Оптимальный вид спорта для людей, страдающих гемофилией, – плавание.
Людям с гемофилией рекомендуется носить на себе медальон с информацией о болезни. Это может спасти жизнь в критической ситуации.
Для профилактики кровотечений больные гемофилией должны избегать применения аспирина и нестероидных противовоспалительных препаратов, поскольку они замедляют тромбоцитарную функцию.
Прививки для больного гемофилией не представляют большой опасности. Более того, в список обязательных входит вакцина против гепатита В. Большинство лекарственных препаратов все же следует принимать перорально из-за опасности кровотечения при внутримышечном введении.
ВАЖНО!
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
![Онкология]()
Рассмотрим заболевания крови (вызывающие изменения в строении и соотношении кровяных телец), для которых существуют нормативные показатели.
Анемия – снижение количества гемоглобина или транспортирующих его частиц – эритроцитов. Следствием этого заболевания является ослабление снабжения кислородом тканей и органов. Анемия может быть обусловлена нарушением формы эритроцитов, снижением их выработки, ускоренным разрушением или недостатком в организме железа и ряда витаминов. Некоторые болезни этой группы носят генетический характер, другие приобретаются в течение жизни. Признаки сниженного гемоглобина: бледность, сонливость, одышка, утомляемость, головные боли и нарушения сердечного ритма.
Полицитемия – выработка чрезмерного количества эритроцитов. Следствием этого становится синхронное повышение содержания и других кровяных телец, что несёт опасность тромбов, поскольку кровь сильно сгущается.
Талассемия – ускоренная гибель эритроцитов, связанная со сбоем в синтезе гемоглобина. Это наследственное заболевание, которое предполагает только симптоматическую медицинскую помощь.
Малярия – поражение красных кровяных телец мельчайшими паразитами. В результате заражения эритроциты начинают лопаться, ухудшается общее состояние, возможны повреждения внутренних органов. Современная медицина обладает эффективными средствами борьбы с малярией.
2. Лейкоциты
Лимфома – заболевание, при котором злокачественные лейкоциты атакуют лимфатическую систему. Бесконтрольно размножаясь, они вытесняют здоровые лейкоциты. При этом сами изменённые клетки крови уже не способны выполнять необходимые функции.
Лейкоз. Рак костного мозга, обусловленный появлением мутации в стволовых клетках. Как следствие, нарушается выработка здоровых лейкоцитов, которые постепенно замещаются раковыми. Различают острое течение (лейкоз) и хроническое (лейкемия).
Лейкопения. Это симптом, характеризующийся снижением содержания лейкоцитов в крови. Лейкопения может носить временный характер и быть реакцией на некоторые внутренние и внешние факторы (инфекции, полученное излучение, дефицит витаминов). Как самостоятельное заболевание встречается редко.
Лейкоцитоз – слишком большое содержание лейкоцитов в крови. Различают физиологическое и патологическое повышение числа белых кровяных телец. Первое возможно при некоторых состояниях, нагрузках, инфекциях.
Патологический лейкоцитоз проявляется при тяжёлых заболеваниях (онкологических, воспалительных, аутоиммунных). При лейкоцитозе наблюдается слабость, подкожные кровоизлияния, боли в конечностях, потеря веса, ухудшение зрения.
Большинство заболеваний, связанных с нарушением числа лейкоцитов, относятся к онкологическим. Лечение проводится путём пересадки костного мозга, химио- и лучевой терапией.
3. Тромбоциты
Тромбоцитопения возникает как следствие болезней костного мозга или из-за слишком интенсивного разрушения тромбоцитов в селезёнке. Недостаток тромбоцитов вызывает кровоточивость слизистых, носовые кровотечения, подкожные кровоизлияния, внутренние кровотечения (что очень опасно). Некоторые заболевания, связанные с понижением числа тромбоцитов, имеют наследственное происхождение.
Тромбоцитоз – чрезмерное количество тромбоцитов в крови. Различают первичный тромбоцитоз (чрезмерная выработка костным мозгом тромбоцитов) и вторичный (реактивное повышение содержания тромбоцитов вследствие инфекций, травм, соматических и инфекционных заболеваний).
4. Плазма крови
Заболевания плазмы крови не менее серьёзны, чем нехватка или переизбыток кровяных телец. Некоторые из них развиваются так стремительно, что больного бывает сложно спасти. Плазма крови нарушается при следующих болезнях:
Сепсис – инфицирование самой крови, призванной бороться с инфекциями в органах и тканях. Сепсис очень опасен, поскольку инфекция охватывает с током крови сразу весь организм.
Гемофилия – наследственное заболевание, которым болеют только мужчины. Отсутствие определённых белков в плазме крови делает кровь почти не способной к свёртыванию. При гемофилии малейшее кровотечение оборачивается большой кровопотерей, которую трудно остановить.
Болезнь фон Виллебранда – белок, необходимый для свёртывания крови, вырабатывается и присутствует в плазме, но он не выполняет свои функции. Это заболевание также опасно кровотечениями.
Множественная миелома. Злокачественное изменение клеток плазмы крови, в результате которого поражаются различные органы и нарушаются важные функции.
Заболевания
![частые заболевания]()
Первичное обследование
![Жалобы и симптомы]()
- Анализы крови на онкомаркеры
- МРТ головы, позвоночника, органов малого таза, органов брюшной полости
- МСКТ органов грудной клетки
- МСКТ органов брюшной полости (виртуальная колоноскопия)
- Гастроскопия (возможность проведения под наркозом, биопсия)
- Колоноскопия (возможность проведения под наркозом, биопсия)
- Бронхоскопия (с биопсией)
- Сцинтиграфия (костей скелета, легких)
Если вы обнаружили у себя подобные симптомы, возможно, это сигнал заболевания, поэтому рекомендуем проконсультироваться с нашим специалистом.
Диагностика
![Диагностика]()
- Анализы крови на онкомаркеры
- МРТ головы, позвоночника, органов малого таза, органов брюшной полости
- МСКТ органов грудной клетки
- МСКТ органов брюшной полости (виртуальная колоноскопия)
- Гастроскопия (возможность проведения под наркозом, биопсия)
- Колоноскопия (возможность проведения под наркозом, биопсия)
- Бронхоскопия (с биопсией)
- Сцинтиграфия (костей скелета, легких)
Наши цены
![Типичные жалобы]()
- Консультация врача онколога, профессор - 10000 р.
- Онкомаркеры:
- ПСА (простатический специфический антиген) общий - 575 р.
- ПСА общий/ПСА своб.(отношение) - 1040 р.
- РЭА (раковый эмбриональный антиген) - 730 р.
- СА 15-3 - 780 р.
- СА 19-9 - 770 р.
- СА 125 - 740 р.
- UBC (антиген рака мочевого пузыря) - 1775 р.
- СА 72-4 - 1170 р.
- Cyfra 21-1 - 1220 р.
- NSE (нейрон-специфическая енолаза) - 1550 р.
- SCC (антиген плоскоклеточной карциномы) - 1550 р.
- НЕ 4 (секреторный белок эпидидимиса 4) - 1150 р.
- СА 242 - 900 р.
- Белок S 100 - 2475 р.
- CgA (хромогранин А) - 2490 р.
- МРТ (головы, позвоночника, органов малого таза. органов брюшной полости) один отдел на базе ЛПУ-партнера - от 6000 р.
- МСКТ органов грудной клетки на базе ЛПУ-партнера - от 6000 р.
- Гастроскопия (ЭГДС, эзофаго-гастро-дуоденоскопия) на базе ЛПУ-партнера без наркоза/с наркозом (во сне) - от 6000/10000 р.
- Колоноскопия на базе ЛПУ-партнера без наркоза/с наркозом (во сне) - от 12000/20000 р.
- ПЭТ-КТ на базе ЛПУ-партнера - от 35000 р.
- Сцинтиграфия костей скелета на базе ЛПУ-партнера - от 8000 р.
Мы стараемся оперативно обновлять данные по ценам, но, во избежание недоразумений, просьба уточнять цены в клинике.
Данный прайс-лист не является офертой. Медицинские услуги предоставляются на основании договора.
Читайте также: