
Что такое мукополисахаридоз кратко
Обновлено: 04.07.2024
Мукополисахаридозы (МПС) — группа наследственных лизосомных нарушений накопления. Лизосомы работают как первичные пищеварительные единицы в клетках. Ферменты в лизосомах расщепляют или переваривают питательные вещества, такие как определенные углеводы и жиры. У людей с МПС, дефицит или сбой лизосомальных ферментов приводит к аномальному накоплению сложных углеводов (мукополисахариды или гликозаминогликаны) в артериях, костях, глазах, суставах, ушах, коже и/или зубах. Скопления гликозаминогликанов могут также обнаруживаться в дыхательной системе, печени, селезенке, центральной нервной системе, крови и костном мозге. Это в конечном итоге вызывает прогрессирующее повреждение клеток, тканей и различных систем организма.
В большинстве случаев мукополисахаридозы — это хронические, прогрессирующие расстройства и, в зависимости от типа МПС и степени тяжести, у пострадавших людей может наблюдаться снижение физической и психической функции, что часто приводит к опасным для жизни осложнениям.
Болезнь возникает в результате дефицита или сбоя в работе лизосомального фермента, необходимого для расщепления дерматансульфата, гепарансульфата или кератансульфата (по отдельности или вместе). Неспособность разрушить мукополисахариды приводит к их накоплению в клетках, тканях и органах по всему организму. Все эти нарушения наследуются как аутосомно-рецессивные признаки, за исключением синдрома Хантера, который сцеплен с Х-хромосомой.
Рецессивные генетические нарушения возникают, когда человек наследует аномальный ген от каждого родителя. Если человек получает один нормальный ген и один ген для заболевания, человек будет носителем заболевания, но симптомы не будут развиваться. Риск для двух родителей-носителей, чтобы передать дефектный ген и, следовательно, иметь больного ребенка, составляет 25% с каждой беременностью. Риск иметь ребенка, который будет носителем, как родители, составляет 50% с каждой беременностью. Шанс для ребенка получить нормальные гены от обоих родителей и быть генетически нормальным при двух родителях-носителях составляет 25%.
Классификация мукополисахаридозов
При такой болезни как мукополисахаридоз типы синдромов отражают выпадение определенного фермента.
Синдром Херлера (мукополисахаридоз типа 1-H) — наиболее тяжелая форма мукополисахаридоза. Он характеризуется дефицитом фермента альфа-L-идуронидазы, что приводит к накоплению сульфатов дерматана и гепарана. Симптомы расстройства начинают проявляться в возрасте от шести месяцев до двух лет. Пострадавшие дети могут испытывать задержки развития, рецидивирующие инфекции мочевыводящих путей и верхних дыхательных путей, шумное дыхание и постоянные выделения из носа. Дополнительные физические проблемы могут включать помутнение роговицы глаза, необычно большой язык, серьезную деформацию позвоночника и скованность суставов. Психическое развитие начинает регрессировать в возрасте около двух лет.
Синдром Шайе (мукополисахаридоз типа IS) — напротив, самая легкая формя болезни. Как и при синдроме Херлера, у людей с синдромом Шейе наблюдается дефицит фермента альфа-L-идуронидазы. Однако при синдроме Шейе дефицит специфичен для накопления только дерматансульфата. Люди с синдромом Шейе имеют нормальный интеллект, рост и продолжительность жизни. Симптомы включают в себя жесткие суставы, синдром запястного канала, обратный кровоток в сердце (регургитацию аорты) и помутнение роговицы, которое может привести к потере остроты зрения. Появление симптомов у людей с синдромом Шейе обычно происходит в возрасте около пяти лет.
Дополнительные симптомы включают:
- низкий рост;
- сердечные нарушения;
- нарушения дыхания;
- увеличение печени и селезенки (гепатоспленомегалия);
- неврологические нарушения.
Тяжесть различных расстройств МПС может изменяться в широких пределах даже среди людей с одинаковым типом болезни.
В большинстве случаев больные младенцы выглядят нормальными при рождении, и симптомы проявляются примерно в возрасте одного или двух лет, однако, при МПС VII приблизительно 40% беременностей с затронутым ребенком осложняются состоянием, называемым неиммунной водянкой плода. Она может быть обнаружена при обычном ультразвуковом исследовании.
Начальные симптомы могут включать частые простуды, насморк, инфекции, задержки роста или умеренные задержки развития.
Легкие формы болезни могут не проявляться до подросткового периода или юности.
Диагностика мукополисахаридоза
Анализ мочи
Основа для скрининговых тестов на МПС — анализ мочи на чрезмерную экскрецию гликозаминогликанов (GAG). В зависимости от того какого типа мукополисахаридоз диагностика мочевых GAG может выявить избыток дерматансульфата, гепарансульфата или двух веществ сразу.
Исследования изображений
Множественный диастаз определяется на основании простой рентгенографии. Это классический диагностический критерий при синдроме Херлера. Эти выводы включают следующее:
- большой череп с преждевременным срастанием шва, j-образным турецким седлом и неглубокими глазницами;
- ненормальное расстояние между зубами с зубными кистами;
- короткие утолщенные и неправильные ключицы;
- короткие, широкие и фаланги в форме трапеции;
- веслообразные ребра;
- передняя гипоплазия поясничных позвонков с кифозом;
- плохо сформированный таз с небольшими головками бедер;
- увеличенные диафизы длинных костей и разноразмерные метафизы.
Лечение мукополисахаридоза
При такой болезни как мукополисахаридоз, лечение, направленное на причину заболевания, пока не разработано. Долгое время не существовало и патогенетического лечения мукополисахаридозов.
Во многих случаях положительный эффект оказывает трансплантация костного мозга. Но проводить ее у таких пациентов сложно, поэтому количество клиник, где такие операции успешно проводятся невелико.
Ферментно-заместительная терапия стала прорывом в патогенетической терапии МПС. Но на территории стран СНГ соответствующие препараты пока малодоступны ввиду отсутствия официальной регистрации.
Терапевтическое лечение
Ферменто-заместительная терапия стала в Бельгии стандартным терапевтическим вариантом для пациентов с некоторыми типами МПС.
Ларонидаза представляет собой полиморфный вариант человеческого фермента альфа-L-идуронидазы, продуцируемого по технологии рекомбинантных ДНК. Она показана для лечения МПС типа I (формы Херлера и Херлера-Шейе). Препарат увеличивает катаболизм гликозаминогликанов (GAG), которые накапливаются с МПС I. Ларонидазная терапия улучшает подвижность суставов и легочную функцию.
Идурсульфаза представляет собой очищенную форму человеческого идуронат-2-сульфатазы, лизосомального фермента. Он гидролизует 2-сульфатные эфиры концевых остатков идуронатсульфата из GAG-дерматансульфата и гепарансульфата в лизосомах различных типов клеток. Он используется для замены недостаточного уровня лизосомального фермента идуронат-2-сульфатазы в МПС II.
Элосульфаза-α одобрена Управлением по контролю за продуктами и лекарствами ЕС (ЕМА) для пациентов с синдромом Моркио А (мукополисахаридоз типа IVA). Исследование, проведенное Hendriksz и соавторами, выявило доказательства того, что длительное использование этого препарата обеспечивает частичное восстановление функциональных способностей у пациентов с этим синдромом.
Вестронидаза-α одобрена ЕМА для МПС VII. Это препарат с высокой степенью эффективности, а большинство его побочных реакций легкой или средней степени тяжести.
Трансплантация костного мозга
Трансплантация костного мозга (ТКМ) была успешной в лечении состояний МПС, особенно синдрома Херлера. Дети, получающие лечение ТКМ, обычно имеют увеличенную продолжительность жизни. Непрооперированные дети обычно умирали от кардиореспираторного компромисса в первом десятилетии жизни. Тем не менее, скелетно-мышечное состояние (мультиплекс дизостоза) не улучшилось с BMT. Рентгенограммы скелета детей, которых лечили BMT, и тех, кто не лечился, обычно выглядят одинаково.
Хирургическая помощь
Пациенты с МПС часто требуют многократных сложных хирургических процедур и имеют высокую послеоперационную смертность из-за респираторных и сердечных заболеваний. Поэтому оказание хирургической помощи таким пациентам в Бельгии проводится под особым контролем многопрофильной команды медицинских специалистов.
Гидроцефалия. Вентрикулоперитонеальное шунтирование - это хирургическое лечение выбора у ребенка с гидроцефалией. Некоторое клиническое улучшение было отмечено у этих пациентов после шунтирования.
Помутнение роговицы. Трансплантация роговицы помогает в этом случае восстановить зрение.
Сердечно-сосудистые заболевания также часто сопровождают мукополисахаридоз — рекомендации в этом случае сводятся к протезированию пораженного митрального или аортального клапана.
Апноэ во сне — обычное явление при МПС и определяется как прекращение воздушного потока через рот или нос на срок более 10-15 секунд. Трахеостомия используется для успешного лечения тяжелого апноэ, особенно для пациентов с атлантоаксиальной нестабильностью при синдроме Моркио.
Ортопедические хирургические процедуры включают операции на мягких тканях и исправление костных аномалий. Наиболее распространенной операцией, выполняемой у этих пациентов, стало высвобождение запястного канала.
Кифоз прогрессирует у многих больных, особенно на уровне грудопоясничного отдела и иногда связан с грудным сколиозом. Задний спондилодез используется для предотвращения дальнейшего прогрессирования.
Осложнения мукополисахаридоза
Заболевание центральной нервной системы
Гидроцефалия обычно наблюдается у этих пациентов. Считается, что она становится результатом дефекта реабсорбции спинномозговой жидкости (CSF). Тяжесть гидроцефалии коррелирует с тяжестью умственной и неврологической отсталости. Миелопатия шейного отдела позвоночника, вторичная по отношению к атлантоаксиальной нестабильности, также наблюдается часто.
Сердечно-сосудистые заболевания
Симптомы сердечных заболеваний присутствуют у многих пациентов. Многие из них имеют симптомы стенокардии, вторичные к атеросклерозу и ишемии. Они также могут проявляться дисфункцией клапанов, гипертонией и застойной сердечной недостаточностью. Возможны внезапный сердечно-сосудистый коллапс и смерть.
Болезнь легких
Обструктивное заболевание дыхательных часто сопровождает МПС. Оно вызвано сужением трахеи и бронхиальных дыхательных путей, утолщенными голосовыми связками и избыточной тканью в верхних дыхательных путях. Эти характеристики могут вызывать проблемы, начиная от апноэ во сне до тяжелой дыхательной недостаточности и легочного сердца.
Мукополисахаридозы оказывают значительное влияние на развитие костно-мышечной системы, в том числе развивается тугоподвижность суставов, уродства и прогрессирующая потеря функции. Тип и степень поражения костно-мышечной системы зависит от типа заболевания. Прогноз тоже определяется в зависимости от типа болезни. У большинства пациентов без патогенетического лечения продолжительность жизни остается короткой, а некоторые умирают в младенчестве.
Трансплантация костного мозга оказывает некоторые положительные системные эффекты, такие как уменьшение гепатоспленомегалии, обструкции дыхательных путей и сердечно-легочных заболеваний. Эти эффекты привели к увеличению продолжительности жизни, поэтому многие из оперированных пациентов выживают после первого десятилетия жизни.
Определяющим фактором длительного выживания должна стать заместительная терапия, но пока опыт ее использования слишком короток, чтобы можно было говорить о продолжительности жизни.
Клиники Бельгии
Статья подготовлена по материалам:
Мукополисахаридозы – это группа генетически обусловленных заболеваний, возникающих вследствие нарушения обмена кислых мукополисахаридов (гликозаминогликанов). Характерны системные поражения скелета и задержка физического развития. При некоторых формах наблюдается умственная отсталость. Возможны нарушения сердечной деятельности, патология органов зрения, образование грыж, неврологические нарушения, гипертрихоз, увеличение печени и селезенки. Диагноз выставляется на основании клинических признаков, данных рентгенографии и других исследований. Лечение симптоматическое.
МКБ-10
Общие сведения
Мукополисахаридозы – группа генетических заболеваний, сопровождающихся накоплением кислых мукополисахаридов в органах и тканях. Причиной развития является передающаяся по наследству неполноценность лизосомных ферментов. Впервые мукополисахаридоз был описан Гурлер в 1917 году. Лечение мукополисахаридоза осуществляют травматологи-ортопеды при участии кардиологов, офтальмологов, неврологов, отоларингологов и других специалистов.
Виды мукополисахаридоза
Мукополисахаридоз типа IH
Мукополисахаридоз типа IH или синдром Гурлер встречается у 1 новорожденного из 20-25 тысяч. Симптомы мукополисахаридоза появляются в течение первого года жизни, полная клиническая картина формируется к 1-2 годам. Для данной формы мукополисахаридоза характерны грубые черты лица и деформация черепа в форме лодочного киля (скароцефалия). Из-за увеличенных аденоидов и пороков развития в области лица и носа больные дышат ртом. Отмечаются прогрессирующие деформации конечностей и других частей скелета, отставание в росте.
Тоны сердца приглушены, границы расширены. При аускультации определяются систолические шумы, на ЭКГ выявляется диффузное поражение миокарда. Передняя брюшная стенка пациентов с мукополисахаридозом ослаблена, живот увеличен, часто выявляются гидроцеле, пупочные и паховые грыжи. Печень и селезенка увеличены. Характерны нарушения зрения и слуха. Возможно помутнение и увеличение роговицы, пигментная дистрофия сетчатки, глаукома, атрофия зрительных нервов и застойные явления в области глазного дна. У больных мукополисахаридозом этого типа развивается прогрессирующая умственная отсталость. Могут возникать нарушения координации движений, парезы и параличи. Выявляется избыточный рост пушковых волос.
Рентгенография позвоночника пациентов с мукополисахаридозом свидетельствует о характерной деформации позвонков. Позвонки имеют кубовидную форму, их контуры закруглены, в переходном отделе выявляется скошенность передневерхних углов, углообразный кифоз, утолщение и укорочение отростков. На рентгенографии грудной клетки определяется утолщение передних и истончение задних отделов ребер, сопровождающиеся их лопатовидной или саблевидной деформацией, укорочение и деформация ключиц, уменьшение и смещение головок плечевых костей.
Рентгенография таза подтверждает сужение тазового кольца, скошенность вертлужных впадин и уменьшение головок бедренных костей. На рентгенограммах трубчатых костей выявляется истончение кортикального слоя и расширение костномозгового канала. Рентгенография костей кисти свидетельствует о недоразвитии ногтевых фаланг, укорочении и расширении пястных костей, проксимальных и средних фаланг. На рентгенограммах черепа определяется недоразвитие костей лицевого черепа, краниостеноз и макроцефалия.
Мукополисахаридоз типа I-S
Мукополисахаридоз типа I-S или болезнь Шейе (описана в 1962 г. американским офтальмологом Шейе) является более поздним, относительно благоприятно протекающим вариантом мукополисахаридоза типа IH (синдрома Гурлер). До 3-6 лет развитие детей соответствует норме. Первым признаком мукополисахаридоза становятся сгибательные контрактуры пальцев рук. В последующем ограничивается разгибания в лучезапястных, локтевых и плечевых суставах. Контрактуры нижних конечностей, как правило, слабо выражены. Полная клиническая картина мукополисахаридоза формируется к началу подросткового возраста.
Пациенты с мукополисахаридозом коренастые, невысокие, с грубыми чертами лица и хорошо развитой мускулатурой. Отмечается повышенное оволосение (гипертрихоз). Часто возникают паховые или пупочные грыжи. Кожа на пальцах натянута и утолщена. Из-за сдавления срединного нерва возможно развитие синдрома запястного канала, сопровождающееся атрофией мышц области тенара и парестезиями в области III-IV пальцев. У некоторых больных мукополисахаридозом выявляется аортальный стеноз, недостаточность клапанов аорты, пигментная дистрофия сетчатки, глаукома и помутнение роговицы. Интеллект в норме, увеличение селезенки и печени нехарактерно. На рентгенограммах определяется картина, аналогичная мукополисахаридозу типа IH, но патологические изменения выражены менее резко.
Мукополисахаридоз типа II
Также при данной форме мукополисахаридоза наблюдается незначительное увеличение селезенки и печени, прогрессирующая тугоухость, узелки на коже спины и некоторое снижение интеллекта. В последующем может развиться помутнение роговицы. Рентгенологическая картина – как при мукополисахаридозе типа I-S. Возможны два варианта развития болезни: благоприятный (вариант В) и неблагоприятный (вариант А). При благоприятном варианте симптомы выражены нерезко, иногда возникает незначительная умственная отсталость, пациенты с мукополисахаридозом могут доживать до 30 и более лет. Для неблагоприятного варианта характерна яркая клиническая картина и серьезные нарушения интеллекта. Летальный исход наступает в подростковом возрасте.
Мукополисахаридоз типа III
Мукополисахаридоз типа III или болезнь Санфилиппо (описана в 1963 г. американским педиатром Санфилиппо) развивается у 1 новорожденного из 100-200 тысяч. В первые годы жизни развитие соответствует возрасту, иногда наблюдаются затруднения глотания и неуклюжая походка. В возрасте 3-5 лет ребенок с мукополисахаридозом становится апатичным и начинает отставать в развитии. Возникают нарушения речи, огрубление черт лица, недержание мочи и кала. Со временем умственная отсталость прогрессирует и занимает центральное положение в клинической картине этого типа мукополисахаридоза.
Наряду с нарушениями интеллекта наблюдается умеренное увеличение печени и селезенки, повышенное оволосение, контрактуры и задержка роста. Патология со стороны глаз и сердечно-сосудистой системы для этой формы мукополисахаридоза нехарактерна. Рентгенологическая картина – без изменений или как при мукополисахаридозе типа I-S, но менее выраженная. Больные мукополисахаридозом третьего типа, как правило, погибают в возрасте 10-20 лет от инфекционных осложнений.
Мукополисахаридоз типа IV
Мукополисахаридоз типа IV или болезнь Моркио (описана в 1929 г. уругвайским педиатром Моркио) наблюдается у 1 новорожденного из 40 тысяч. До 1-3 лет дети развиваются нормально. В последующем возникает значительное отставание в росте, укорочение шеи и туловища, контрактуры и вальгусная деформация конечностей, сколиоз или кифоз, разнообразные деформации грудной клетки, снижение силы мышц, утолщение кожи и огрубление черт лица. При этом типе мукополисахаридоза часто развиваются паховые и пупочные грыжи, дистрофия роговицы и тугоухость. Интеллект сохранен.
На рентгенограммах позвоночника определяется кифоз, сколиоз, расширение и уплощение тел позвонков. При проведении рентгенографии таза и конечностей выявляются множественные деформации, неровность контуров, уплощение головок бедренных костей, укорочение костей предплечья и деформации стоп. Средняя продолжительность жизни больных мукополисахаридозом – менее 20 лет. Смерть при данной форме мукополисахаридоза наступает из-за сопутствующих заболеваний, осложняющихся сердечно-легочной недостаточностью.
Другие типы мукополисахаридоза
Мукополисахаридоз типа VI или болезнь Марото-Лами (описана в 1960 г. французами Лами и Марото) развивается в возрасте 2 года и старше. Возникает огрубление черт лица, отставание в росте, укорочение шеи, контрактуры суставов и бочкообразная деформация грудной клетки. Характерны частые простуды. Возможны грыжи, увеличение печени и селезенки. Интеллект не страдает. Наблюдается два варианта течения: классический и мукополисахаридоз со слабой выраженностью клинических проявлений. На рентгенограммах больных мукополисахаридозом определяется кубовидная или клиновидная деформация позвонков, треугольная деформация таза, недоразвитие и деформация головок бедренных костей, укорочение малоберцовых костей.
Мукополисахаридоз типа VII протекает, как мупоколисахаридоз типа III, различия выявляются только при проведении биохимических исследований. Мукополисахаридоз типа VIII по симптомам напоминает мукополисахаридоз типа IV, но, в отличие от него, сопровождается задержкой умственного развития.
Диагностика
Диагноз мукополисахаридоза устанавливается на основании характерной клинической и рентгенологической картины, выявления гликозаминогликанов в моче, изучения активности ферментов в клеточных культурах, генетического секвенирования. В ходе обследования больным мукополисахаридозом назначаются консультации различных специалистов: кардиолога, гастроэнтеролога, офтальмолога, отоларинголога, невролога, психиатра и т. д., проводятся инструментальные исследования для оценки состояния различных органов и систем.
Лечение мукополисахаридоза
Патогенетическая терапия не разработана. Лечение симптоматическое, может быть как консервативным, так и оперативным. Осуществляется профилактика и лечение респираторных инфекций, проводится коррекция нарушений зрения и слуха. При необходимости выполняются грыжесечение и герниопластика, операции по устранению контрактур и коррекции деформаций скелета.
Прогноз и профилактика
Прогноз при всех типах мукополисахаридоза неблагоприятный – продолжающееся накопление продуктов обмена в тканях приводит к усугублению патологических изменений со стороны всех органов и систем. Использование любых лечебных средств (переливания крови, введение гормонов и т. д.) при мукополисахаридозе обеспечивает лишь временное улучшение. Рекомендуется пренатальная профилактика.
1. Руководство по педиатрии. Врожденные и наследственные заболевания/ Новиков П.В., Баранов А.А., Каганов Б.С., Шиляев Р.Р - 2007
Патологические изменения, лежащие в основе заболевания
Мукополисахаридозы представляют собой группу из 11 редких наследственных (т.е. передающихся от родителей к детям) заболеваний, которые связаны с нарушением обмена гликозаминогликанов. Данные вещества присутствуют в организме человека повсеместно и заполняют пространство между клетками, поэтому нарушение их метаболизма приводит к серьезным нарушениям функций практически каждого органа. В зависимости от гена, который поражается при мукополисахаридозе, определяется тип заболевания. Всего выделяют 11 генов, поломки (мутации) в которых приводят к снижению или потере функции определенных ферментов, разрушающих гликозаминогликаны и приводящих к развитию мукополисахаридоза какого-либо типа. В настоящее время только 4 из 11 типов мукополисахаридозов поддаются лечению: мукополисахаридоз I, II, IV и VI типов. Своевременно начатая терапия позволяет улучшить качество жизни заболевшего и предотвратить серьезные нарушения функций организма.
Мукополисахаридоз I типа (МПС I) – как и другие типы мукополисахаридозов, является наследственным заболеванием, причиной которого является унаследованный от обоих родителей (в большинстве случаев, здоровых) поломанные гены. Из-за генных изменений возникает дефицит фермента (альфа-L-идуронидаза), приводящий к накоплению мукополисахаридов (или гликозаминогликанов) в соединительной ткани всех органов – в центральной нервной системе, сердце, сосудах, опорно-двигательном аппарате, печени, селезенке, органах зрения и слуха.

В зависимости от проявлений заболевания выделяют разные по степени тяжести формы МПС I (см. Признаки и симптомы мукополисахаридоза) 10
Клиническая картина заболевания
Поскольку гликозаминогликаны накапливаются практически во всех органах и тканях, заболевание имеет множество клинических появлений. Наиболее часто родители обращаются к врачу в связи с одним или несколькими проявлениями мукополисахаридоза у ребенка:
Мукополисахаридозы являются врожденными нарушениями метаболизма, вызванными дефицитом лизосомальной глюкозидазы или сульфатазы, который приводит к накоплению мукополисахаридов или гликозоаминогликанов в лизосомах. В таблице ниже приведены основные характеристики заболеваний данной группы.
Данные нарушения будут рассмотрены кратко, так как неврологические проявления часто отодвигаются на задний план дисморфическими и висцеральными проявлениями. Подробности можно узнать в специализированной литературе.
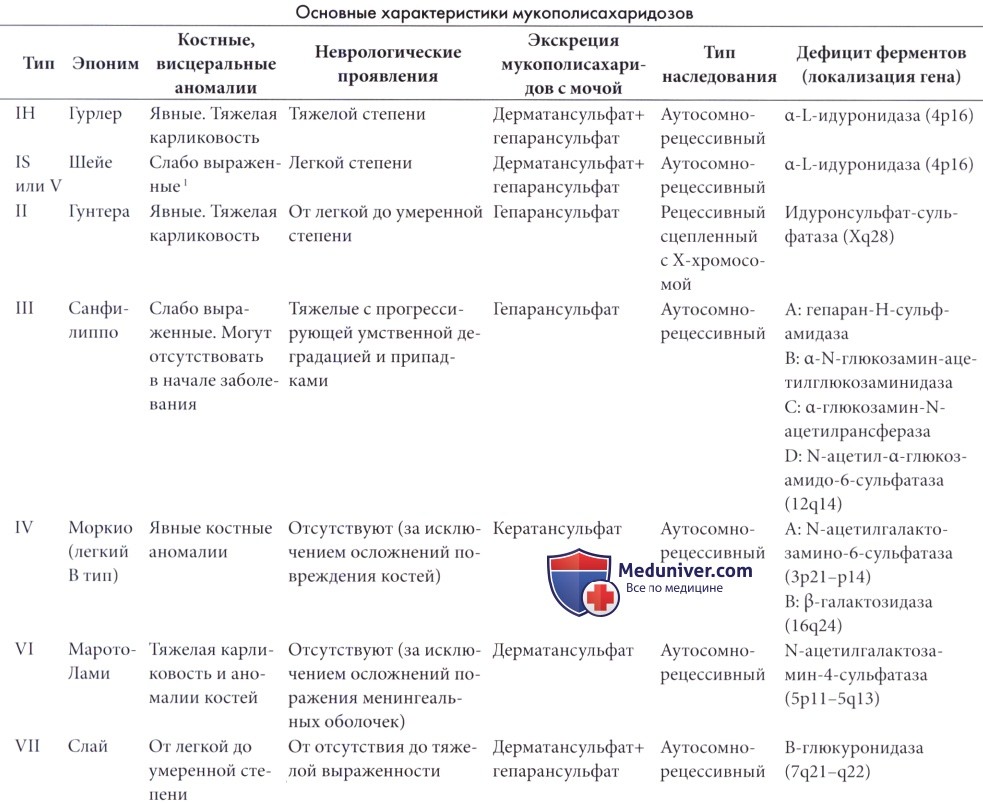
а) Болезни Гурлер и Шейе (мукополисахаридозы IH и 1-H/S типов). При болезни Гурлер широко распространены поражения внутренних органов, а клетки, содержащие полисахариды, обнаруживаются в ретикулоэндотелиальной системе и соединительной ткани.
В головном мозге накопления представлены в основном GM2 и GM3 ганглиозидами, в то время как в периферических тканях основные скопления состоят из сульфата дерматана и гепарана, в большом количестве экскретирующихся с мочой.
Болезнь Гурлер является аутосомно-рецессивным заболеванием, которое встречается с частотой 1 на 100000 новорожденных. Больные дети кажутся нормальными при рождении, но развиваются медленно, костные изменения становятся явными в возрасте от шести месяцев до двух лет.
Выявляется гепатоспленомегалия, отделяемое из носа и пупочная грыжа. Характерные скелетные аномалии формируются постепенно вместе с огрублением черт лица, кифосколиозом, контрактурами суставов и расстройством интеллекта. Отмечаются помутнения роговицы, постоянным признаком является выраженная карликовость.
Голова большая, часто долихоцефалической формы. На КТ и МРТ выявляется расширение желудочков и пониженная плотность белого вещества полушарий (Zarifi et al., 2001; Matheus et al., 2004).
В области гипоталамуса часто выявляется кистозный арахноидит, вызывающий гидроцефалию. Расширение пространств Вирхова-Робена часто хорошо заметно на МРТ. Смерть наступает в возрасте до 20 лет.
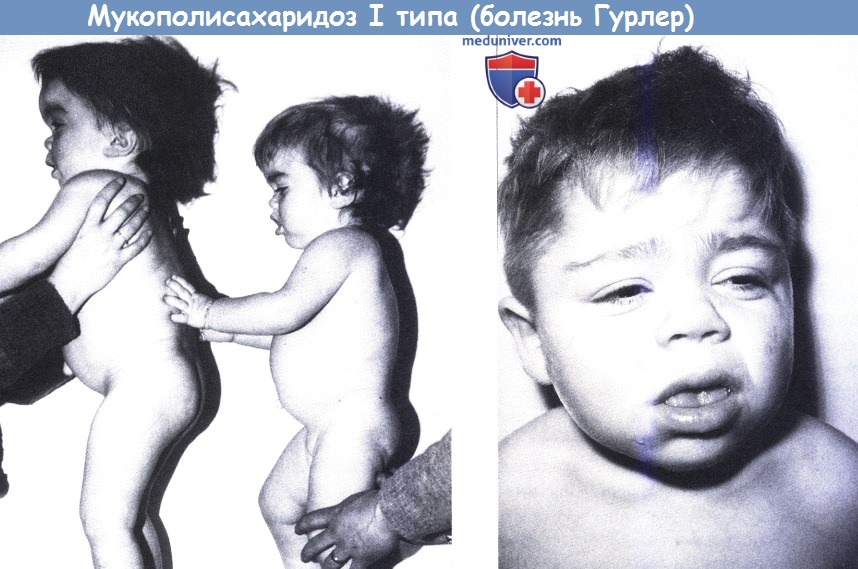
Мукополисахаридоз I типа (болезнь Гурлер).
Слева: Две сестры в возрасте 7 и 3 лет, отмечается увеличение головы со скафокефалией, типичный профиль и короткие кисти с широкими, частично согнутыми пальцами.
Справа: 12-летний мальчик с типичным лицом.
Диагноз может быть подтвержден L-идуронидазным исследованием. Наличие азурофильных гранул в гранулоцитах, гранул Рейли, вакуолизированных лимфоцитов (клетки Гассера I типа) и базофильных клеток в костном мозге (клетки Гассера II типа) является признаком, подтверждающим наличие заболевания. Возможна пренатальная диагностика.
Ген расположен на участке 4р16.3. Известно множество мутаций, две из которых являются причиной 80% случаев заболевания.
Лечение болезни Гурлер в настоящее время основано на ферментозаместительной терапии L-идуронидазой; данный метод является существенной заменой трансплантации костного мозга, которая была первым методом лечения с частичным успехом.
Ферментозаместительная терапия оказалась хорошо переносимой и эффективной в отношении висцеральных проявлений заболевания. Помутнения роговицы исчезают, а воздействие заболевания на рост и развитие принимает частичный характер. Раннее лечение необходимо до момента формирования стойких аномалий.
Уменьшения выраженности неврологических проявлений не отмечалось, тем не менее, описаны казуистические случаи повышения активности и восприимчивости у детей при раннем лечении (Wraith, 2005). Ограниченная эффективность объясняется неспособностью ферментов проникать через гематоэнцефалический барьер.
Попытки обойти барьер с помощью трансплантации гемопоэтических клеток или стволовых гемопоэтических клеток находятся на стадии изучения (Grewal et al., 2005; Lucke et al., 2007). В целом отмечается тенденция замещения трансплантации костного мозга ферментозаместительной терапией не только при мукополисахаридозах, но и при ряде других лизосомальных болезней накопления (Germain, 2005; Hoffmann и Mayatepek, 2005).
Данный метод лечения используется в основном при тяжелых состояниях с поражением центральной нервной системы, особенно при мукополисахаридозах I, II, III и VI типов. Предстоит решение ряда проблем, в частности, прохождения крупных молекул ферментов через гематоэнцефалический барьер. Изучаются и другие методы лечения, например, генная терапия.
Болезнь Гурлер-Шейе, официально известная как мукополисахаридоз V типа, характеризуется клинической картиной аналогичной болезни Гурлер и возможным заметным дисморфизмом (Schmidt et al., 1987), но поражение нервной системы ограничивается высокой частотой синдрома запястного канала, а умственные способности не изменяются. У некоторых пациентов развивается гидроцефалия.
б) Болезнь Гунтера (мукополисахаридоз II типа). Данное заболевание связано с дефицитом идуронат-2-сульфатазы (Kato et al., 2005) и наследуется сцепленно с Х-хромосомой. Заболевание встречается приблизительно в 5 раз реже, чем мукополисахаридозы I типа. В зависимости от наличия задержки умственного развития выделяются легкие и тяжелые формы. Неврологические аномалии при легких формах заболевания могут включать нейросенсорную тугоухость, пигментный ретинит и умеренную гидроцефалию (Froissart et al., 2002).
В результате утолщения соединительной ткани часто встречаются синдромы ущемления нервов. Определение уровня ферментов обеспечивает антенатальную и постнатальную диагностику. Ген расположен на участке Xq28, в 30% случаев отмечается его делеция. В тяжелых случаях возможно лечение с помощью трансплантации костного мозга.
в) Болезнь Санфипиппо (мукополисахаридоз III типа). Болезнь Санфилиппо является генетически гетерогенным заболеванием, известно четыре типа в зависимости от химического дефекта (табл. 8.1). Клинически заболевание проявляется прогрессирующей задержкой неврологического и умственного развития с началом в возрасте 2-6 лет. Помутнения роговицы отсутствуют, тем не менее, рано появляются аномально грубые черты лица и утолщение волос с последующим формированием неврологических нарушений. Часто встречается эпилепсия.
Течение заболевания неуклонно прогрессирующее, большинство пациентов умирает до 20 лет.
На начальных этапах диагностика затруднена в связи с преобладанием неврологических и психиатрических проявлений. Пациентам с необъяснимым регрессом показан плановый скрининг мочи на экскрецию му-кополисахаридов. Ферментное исследование позволяет проводить дифференцировку различных типов заболевания, так как фенотипические различия отсутствуют. Возможна пренатальная диагностика. Предпринимались попытки лечения с помощью трансплантации костного мозга, стволовых клеток и генной терапии, но результаты были в лучшем случае сомнительными.
г) Болезнь Моркио (мукополисахаридоз IV типа). Болезнь Моркио представлена двумя различными типами в зависимости от генетического и ферментативного дефекта. Клинические проявления представлены в основном костными аномалиями и помутнением роговицы без задержки умственного развития. При А типе заболевания постоянным признаком является гипоплазия зуба второго шейного позвонка, может отмечаться бульбоспинальная компрессия (Montano et al., 2003). Возможно повреждение менингеальных оболочек и нейронов. При легком течении заболевания или В типе неврологические проявления отсутствуют. Часто встречается атлантоаксиальная нестабильность, которая может быть причиной компрессии спинного мозга. Необходимо наблюдение и лечение у ортопеда и невропатолога.
е) Болезнь Слая (мукополисахаридоз VII типа). Болезнь Слая является очень редким заболевание, которое может сопровождаться различными фенотипическими проявлениями от неиммунной водянки плода до случаев, имитирующих болезнь Гурлер с тяжелой задержкой неврологического развития, и случаев с ограниченными проявлениями без неврологических изменений (Bernsen et al., 1987; Saxonhouse et al., 2003).
ж) Другие формы мукополисахаридозов. Атипичные и неклассифицированные формы встречаются редко. Описан новый тип заболевания (Maroteaux, 1973) с основным проявлением в виде ацетоза в сочетании с экскрецией кератансульфата.
Читайте также:
- Что такое диспергирование в химии кратко
- В каких случаях к травмированному месту прикладывают лед обж 8 класс кратко
- Положение о календарном плане воспитательной работы в школе
- Если ребенок не ходил в детский сад как это повлияет на развитие в социуме
- Подумайте используя приведенный в конце урока документ почему испанский философ ортега кратко