
Абсорбционный спектральный анализ кратко
Обновлено: 05.07.2024
Число атомов в возбужденном состоянии не превышает 1-2% от общего числа атомов определяемого элемента в пробе, поэтому аналитический сигнал в атомно-абсорбционной спектроскопии оказывается связанным с существенно большим числом атомов, чем в эмиссионной спектроскопии, и, следовательно, в меньшей степени подвержен влиянию случайных колебаний при работе атомно-абсорбционного спектрофотометра.
Уменьшение интенсивности резонансного излучения в условиях атомно-абсорбционной спектроскопии подчиняется экспоненциальному закону убывания интенсивности в зависимости от длины оптического пути и концентрации вещества, аналогичному закону Бугера-Ламберта-Бера. Если I0 - интенсивность падающего монохроматического света, а I - интенсивность этого света, прошедшего через пламя, то величину lg(I0/I) можно назвать оптической плотностью. Концентрационная зависимость оптической плотности выражается уравнением
lg (I0/I) = А = k l c
где k - коэффициент поглощения; l - толщина светопоглощаюшего слоя (пламени); с - концентрация.
В практике атомно-абсорбционного анализа для количественных определений обычно применяют метод градуировочного графика и метод добавок.
Методы атомно-абсорбционной спектроскопии могут быть использованы или используются в анализе практически любого технического или природного объекта, особенно там, где необходимо определить небольшие содержания элементов. Методики атомно-абсорбционного определения разработаны более чем для 70 элементов периодической системы Д.И. Менделеева.
Предел обнаружения с помощью атомно-абсорбционного анализа для многих элементов характеризуется величиной порядка 10-5. 10-6%. Погрешность определения обычно составляет примерно 5% и в зависимости от различных условий изменяется в пределах от 3 до 10%.
Метод имеет также ряд ограничений. Атомно-абсорбционным методом не определяются элементы, резонансные линии которых лежат в далеком ультрафиолете (углерод, фосфор, галогены и др.).
2.2 Аппаратурное оснащение для осуществления метода
Атомно-абсорбционный спектрофотометр , укомплектованный горелкой для воздушно-ацетиленового пламени, корректором фонового поглощения и источниками резонансного излучения свинца, кадмия, меди, цинка и железа (лампами с полым катодом, безэлектродными разрядными лампами или другими равноценными источниками). Допускается применение спектрофотометра без корректора фонового поглощения при условии проведения экстракционного концентрирования ТУ 4434-009-2990357-95.
2.3 Особенности метода атомно-абсорбционной спектрометрии
Перевод анализируемого объекта в атомизированное состояние и формирование поглощающего слоя пара определенной и воспроизводимой формы осуществляется в атомизаторе - обычно в пламени или трубчатой печи. Наиболее часто используют пламя смесей ацетилена с воздухом (макс. температура 2000°С) и ацетилена с N2O (2700°С). Горелку со щелевидным соплом длиной 50-100 мм и шириной 0,5-0,8 мм устанавливают вдоль оптической оси прибора для увеличения длины поглощающего слоя.
Трубчатые печи сопротивления изготавливают чаще всего из плотных сортов графита. Для исключения диффузии паров через стенки и увеличения долговечности графитовые трубки покрывают слоем газонепроницаемого пироуглерода. Максимальная температура нагрева достигает 3000 °С. Менее распространены тонкостенные трубчатые печи из тугоплавких металлов (W, Та, Мо), кварца с нихромовым нагревателем.
Для защиты графитовых и металлических печей от обгорания на воздухе их помещают в полугерметичные или герметичные камеры, через которые продувают инертный газ (Аr, N2).
Введение проб в поглощающую зону пламени или печи осуществляют разными приемами. Растворы распыляют (обычно в пламя) с помощью пневматических распылителей, реже - ультразвуковых. Первые проще и стабильнее в работе, хотя уступают последним в степени дисперсности образующегося аэрозоля. Лишь 5-15% наиболее мелких капель аэрозоля поступает в пламя, а остальная часть отсеивается в смесительной камере и выводится в сток. Максимальная концентрация твердого вещества в растворе обычно не превышает 1%. В противном случае происходит интенсивное отложение солей в сопле горелки.
Термическое испарение сухих остатков растворов - основной способ введения проб в трубчатые печи. При этом чаще всего пробы испаряют с внутренней поверхности печи; раствор пробы (объемом 5-50 мкл) вводят с помощью микропипетки через дозировочное отверстие в стенке трубки и высушивают при 100°С. Однако пробы испаряются со стенок при непрерывном возрастании температуры поглощающего слоя, что обусловливает нестабильность результатов. Чтобы обеспечить постоянство температуры печи в момент испарения, пробу вводят в предварительно нагретую печь, используя угольный электрод (графитовую кювету) графитовый тигель (печь Вудриффа), металлический или графитовый зонд. Пробу можно испарять с платформы (графитового корытца), которую устанавливают в центре печи под дозировочным отверстием. В результате значительного отставания температуры платформы от температуры печи, нагреваемой со скоростью около 2000 К/с, испарение происходит при достижении печью практически постоянной температуры.
Для введения в пламя твердых веществ или сухих остатков растворов используют стержни, нити, лодочки, тигли из графита или тугоплавких металлов, помещаемые ниже оптической оси прибора, так что пары пробы поступают в поглощающую зону с потоком газав пламени. Графитовые испарители в ряде случаев дополнительно подогревают электрическим током. Для исключения механических потерь порошкообразных проб в процессе нагрева применяются испарители типа цилиндрических капсул, изготовленные из пористых сортов графита.
Иногда растворы проб подвергают в реакционном сосуде обработке в присутствии восстановителей, чаще всего NaBH4. При этом Hg, например, отгоняется в элементном виде, As, Sb, Bi и других - в виде гидридов, которые вносятся в атомизатор потоком инертного газ.. Для монохроматизации излучения используют призмы или дифракционные решетки; при этом достигают разрешения от 0,04 до 0,4 нм.
При атомно-абсорбционном анализе необходимо исключить наложение излучения атомизатора на излучение источника света, учесть возможное изменение яркости последнего, спектральные помехи в атомизаторе, вызванные частичным рассеянием и поглощением света твердыми частицами и молекулами посторонних компонентов пробы. Для этого пользуются различными приемами, например модулируют излучение источника с частотой, на которую настраивают приемно-регистрирующее устройство, применяют двухлучевую схему или оптическую схему с двумя источниками света (с дискретным и непрерывным спектрами). Наиболее эффективна схема, основанная на зеемановском расщеплении и поляризации спектральных линий в атомизаторе. В этом случае через поглощающий слой пропускают свет, поляризованный перпендикулярно магнитному полю, что позволяет учесть неселективные спектральные помехи, достигающие значений А = 2, при измерении сигналов, которые в сотни раз слабее.
Методы атомно-абсорбционного анализа применяют также для измерения некоторых физических и физико-химических величин - коэффициент диффузии атомов в газах, температур газовой среды, теплот испарения элементов и др.; для изучения спектров молекул, исследования процессов, связанных с испарением и диссоциацией соединений.
3 Методика выполнения измерений массовой доли свинца в мясных консервах для детского питания методом атомно-абсорбционной спектрометрии [1,2].
3.1 Подготовка к выполнению измерений
3.1.1 Подготовка лабораторной посуды
Новую и сильно загрязненную лабораторную посуду после обычной мойки в растворе любого моющего средства промывают водопроводной и ополаскивают дистиллированной водой. Процедура очистки лабораторной посуды непосредственно перед использованием включает следующие последовательные этапы: мойка посуды горячей азотной кислотой (1:1) по объему, ополаскивание дистиллированной водой, мойка горячей соляной кислотой (1:1) по объему, ополаскивание дистиллированной водой 3-4 раза, ополаскивание бидистиллированной водой 1-2 раза, сушка.
3.1.2 Приготовление стандартных растворов
3.1.2.1 Основные стандартные растворы элементов готовят: для свинца по ГОСТ 26932. Допускается использование готовых коммерческих растворов с гарантированной концентрацией элементов 1000 мкг/см на азотнокислой или солянокислой основе с массовой долей кислоты не менее 1%.
3.1.2.2 Промежуточные стандартные растворы элементов готовят последовательным разбавлением основных растворов в 10 и 100 раз раствором азотной кислоты массовой долей 1%. Эти растворы хранят в герметичной посуде не более года.
3.1.2.3 Стандартные растворы сравнения готовят из промежуточных растворов путем их разбавления тем же раствором кислоты, что и растворы проб. Содержание элементов в испытуемых и стандартных растворах не должно выходить за пределы следующих рабочих диапазонов: для свинца 0,1-2,0 мкг/см. Измерение абсорбции контрольных растворов допускается проводить при содержании элементов ниже указанных пределов. В рабочих диапазонах достаточно иметь по 3-4 раствора сравнения. Растворы концентрацией металлов от 1 до 10 мкг/см хранят не более месяца, концентрацией менее 1 мкг/см готовят ежедневно.
3.1.2.4 В качестве нулевого стандарта применяется раствор азотной или соляной кислоты с массовой долей 1%, используемый для растворения проб и разбавления стандартных растворов сравнения в данной серии испытаний.
3.1.3 Приготовление испытуемого раствора
3.1.3.1 При использовании способа сухого озоления или кислотной экстракции созолением золу растворяют в тигле при нагревании в азотной кислоте (1:1) по объему из расчета 1-5 см кислоты на навеску в зависимости от зольности продукта. Раствор выпаривают до влажных солей. Осадок растворяют в 15-20 см азотной кислоты массовой долей 1%, количественно переносят в мерную колбу вместимостью 25 см и доводят до метки той же кислотой. При неполном растворении золы полученный раствор с осадком упаривают до влажных солей, перерастворяют в минимальном объеме соляной кислоты (1:1) по объему, еще раз упаривают до влажных солей и растворяют в 15-20 см соляной кислоты массовой долей 1%. Раствор количественно переносят в мерную колбу вместимостью 25 см и доводят до метки той же кислотой.
При неполном растворении золы полученный раствор с осадком доводят до объема 30-40 см соляной кислотой с массовой долей 1% и подогревают на водяной бане или электроплитке при слабом нагреве в течение 0,5 ч. Если и в этом случае полного растворения не наблюдается, раствор отфильтровывают через промытый растворителем фильтр, осадок промывают и отбрасывают, а фильтрат переносят в мерную колбу вместимостью 50 см и доводят до метки той же кислотой
3.1.3.2 При использовании способа мокрой минерализации полученный раствор минерализата упаривают до влажных солей и продолжают растворение.
3.1.4 Приготовление контрольного раствора
Контрольные чаши (стаканы, колбы), полученные вместе с минерализатами проб, проводят через все стадии приготовления испытуемых растворов с добавлением тех же количеств реактивов.
3.1.5 Разбавление растворов
Если содержание элемента в испытуемом растворе при измерениях оказывается выше верхнего предела диапазона рабочих содержаний (5.2.3), то проводится разбавление испытуемого раствора нулевым стандартом. Коэффициент разбавления выбирают таким образом, чтобы содержание элемента в разбавленном растворе находилось в середине рабочего диапазона (для меди, цинка и железа в интервале примерно от 1 до 3 мкг/см). Коэффициент разбавления равен
где - объем аликвоты, взятый для разбавления, см3;
У - объем разбавленного раствора, см3.
3.1.6 Экстракционное концентрирование
Концентрирование методом экстракции проводят, если:
а) после предварительных измерений концентрация свинца в исходном растворе оказалась ниже 0,1мкг/см;
б) имеется необходимость повышения точности анализа;
в) содержание элемента в исходном растворе оказывается ниже достигнутого в данной серии измерений предела обнаружения и имеется необходимость двусторонней оценки содержания элемента в продукте;
г) при определении свинца не проводится коррекция фонового поглощения.
В стаканы вместимостью 100 или 150 см помещают аликвоты испытуемых растворов объемом 10-50 см в зависимости от требований к степени концентрирования и такие же по объему аликвоты контрольных растворов и доводят их объем до 50 см нулевым стандартом. Коэффициент разбавления этих растворов учитывается, как в 5.5. Одновременно в такие же стаканы помещают по 50 см стандартных растворов сравнения.
При проведении экстракции с целью повышения чувствительности и точности анализа используют раствор сравнения с минимальной концентрацией, полученный по 2.3, стандартные растворы с содержанием элемента в 2 и 10 раз ниже минимальной и нулевой стандарт, полученный по 2.4.
При использовании спектрофотометров, не имеющих корректоров фонового поглощения, концентрация элементов в растворах сравнения, взятых для экстракции, не должна превышать следующих уровней: для свинца - 2 мкг/см, для кадмия - 0,1 мкг/см. В стаканы приливают по 10 см раствора лимонной кислоты, добавляют по 2-3 капли раствора фенолфталеина и титруют раствором аммиака до появления слабо-розовой окраски. Растворы переносят в делительные воронки или мерные колбы вместимостью 100 см, приливают по 5 см раствора диэтилдитиокарбамата натрия и по 5 см эфира и встряхивают в течение 1 мин.
При использовании делительных воронок после разделения фаз нижний водный слой отбрасывают, а органические экстракты собирают в пробирки и закрывают пробками. При проведении экстракции в мерных колбах в них доливают такое количество бидистиллированной воды, чтобы органический слой оказался в горле колбы, и при измерениях отбирают органическую фазу подающим капилляром распылителя непосредственно из горла колбы, не допуская его погружения в водную фазу.
На рассеянном свету экстракты устойчивы в течение рабочего дня
Коэффициент концентрирования равен
где - объем аликвоты, взятый для концентрирования, см;
- объем органической фазы, =5, см3.
3.1.7 Подготовка спектрофотометра к работе и выбор условий измерения
а) прогрева источника резонансного излучения перед началом измерений до получения стабильной интенсивности излучения, но не менее 0,5 ч;
б) юстировки источников резонансного и нерезонансного излучения;
в) прогрева включенной горелки перед началом измерений с одновременной ее промывкой дистиллированной водой в течение 5-10 мин;
г) точной настройки монохроматора на резонансную линию по максимуму излучения при минимальной щели, но проведение измерений при максимальной щели монохроматора;
д) юстировки высоты горелки и соотношения воздух/ацетилен перед каждой серией измерений по максимуму абсорбции одного из стандартных растворов сравнения. Используются наиболее чувствительные линии поглощения элементов со следующими длинами волн: для свинца - 283,3 или 217 нм. Выбор резонансной линии свинца зависит от технических характеристик лампы и спектрофотометра и проводится для данного прибора и лампы по критерию большего отношения сигнал/шум и по меньшему значению дрейфа чувствительности и нулевой линии.
3.2 Проведение измерений
3.2.1 Распыляя в пламя нулевой стандарт (при использовании концентрирования - его экстракт), устанавливают показания прибора на нуль. Затем в порядке возрастания концентрации измеряют абсорбцию стандартных растворов сравнения (или их экстрактов). В конце градуировки отмечают положение нулевой линии при распылении нулевого стандарта.
3.2.2 Измеряют абсорбцию небольшого числа (5-10) испытуемых и контрольных растворов, промывая после каждого измерения систему распылителя и горелки дистиллированной водой или нулевым стандартом (для экстрактов - эфиром) до возвращения сигнала к показаниям, близким к нулю. Повторяют точное измерение абсорбции нулевого стандарта и одного из стандартов сравнения, наиболее близкого по концентрации к испытуемым растворам. Если при этом не отмечается заметного смещения нулевой линии и изменения абсорбции стандарта, продолжают измерения абсорбции испытуемых растворов, периодически повторяя контроль дрейфа нуля и чувствительности и заканчивая измерения полной градуировкой.
Измерение абсорбции каждого раствора проводится не менее 2 раз.
3.2.3 Если в процессе измерений отмечается смещение нулевой линии или изменение чувствительности, каждую малую серию испытуемых растворов измеряют дважды в прямом и обратном порядке последовательности, начиная и заканчивая полной градуировкой. Объем серий определяется скоростью дрейфа: число растворов в серии должно быть таким, чтобы изменение абсорбции стандартов сравнения в последовательных градуировках не превышало 5% отн. Если смещение нулевой линии не корректируется автоматическими устройствами, оно должно учитываться путем введения поправок к сигналам поглощения проб и стандартов. Дрейф нуля внутри каждой малой серии измерений считают линейным.
3.2.4 Определение предела обнаружения
После окончания измерений абсорбции полной серии испытуемых растворов проводят 20-кратное измерение абсорбции стандартного раствора с минимальной концентрацией или любого испытуемого раствора, или смеси остатков растворов с низкой концентрацией элемента. В зависимости от наличия дрейфа измерения проводят по 6.2 или по 6.3 - по той же методике, что и для испытуемых растворов. На основе полученной статистики рассчитывают стандартное (среднее квадратическое) отклонение от среднего значения для единичного измерения , мкг/см. Утроенное значение стандартного отклонения 3 считается пределом обнаружения элемента в растворе при =0,99.
Если в проведенной серии измерений присутствует не менее 10 растворов с концентрацией элемента ниже 0,2 мкг/см, специальные измерения не проводят, а рассчитывают стандартное отклонение по формуле
где - расхождение параллельных измерений концентрации элемента в растворе; - количество растворов.
3.3 Обработка результатов
3.3.1 При наличии в приборе компьютерной системы расчета концентрации по значению абсорбции используют рекомендованные в технической инструкции прибора компьютерные программы. При ручной обработке данных строят график зависимости абсорбции от концентрации. Допускается применять линейную, кусочно-линейную или сглаженную нелинейную аппроксимацию градуировочной функции. Для каждой малой серии измерений при построении графика используют средние арифметические значения абсорбции стандартных растворов сравнения, полученные в двух градуировках (до и после измерений абсорбции испытуемых растворов) и исправленные на значение смещения нулевой линии. По графику определяют концентрацию элемента в испытуемых и контрольных растворах. Значения концентрации, более низкие, чем достигнутый предел обнаружения 3, считаются равными нулю. В расчетах используют средние арифметические значения параллельных измерений.
3.3.2 Если разность оказывается меньше предела обнаружения 3 , то дается односторонняя оценка максимально возможной концентрации элемента в продукте
где - число параллельных измерений абсорбции испытуемого раствора.
3.3.3 За окончательный результат измерений принимают среднее арифметическое результатов двух параллельных определений. Окончательный результат округляют до второго десятичного знака.
3.3.4 Допускаемое расхождение между двумя параллельными результатами полученными в одной лаборатории в одной серии измерений (сходимость ), зависит от массовой доли элемента в продукте и при 0,95 не должно превышать значений.
Метод атомно-абсорбционного анализа (AAA) основан на резонансном поглощении света свободными атомами, возникающем при пропускании пучка света через слой атомного пара. Селективно поглощая свет на частоте резонансного перехода, атомы переходят из основного состояния в возбужденное, а интенсивность проходящего пучка света на этой частоте экспоненциально убывает по закону Бугера-Ламберта:
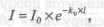
где кн — коэффициент поглощения света;
I — толщина поглощающего слоя.
При практических измерениях обычно пользуются значением оптической плотности поглощения (поглощательной способностью):
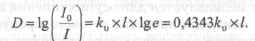
Для применения этого соотношения в количественном химическом анализе необходимо установить связь между коэффициентом поглощения к0 и концентрацией атомов в поглощающем слое.
В современной технике атомно-абсорбционного анализа используются два способа атомизации — атомизация в пламени и электрических атомизаторах.
Атомизация в пламени
Для получения пламени используют различные комбинации горючих газов с окислителями, например водорода, пропана или ацетилена с воздухом или оксидом азота. Кислород в чистом виде почти не применяют как окислитель, так как смеси горючих газов с ним обладают очень высокой скоростью горения, с трудом поддаются контролю.
В практике атомно-абсорбционного анализа наибольшее применение получили два пламени: воздушно-ацетиленовое и пламя оксида азота с ацетиленом. Эти две газовые смеси взаимно дополняют друг друга и совместно позволяют определять примерно 70 элементов. Воздушно-пропановое пламя пригодно в основном для определения щелочных металлов; кадмия, меди, свинца, серебра и цинка.
Первичная реакционная зона (поз. 1, рис. 2.6) для анализа не используется, так как температура в ней менее 1000°С.
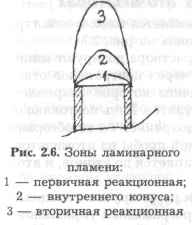
Зона внутреннего конуса (2) благоприятна для измерения атомной абсорбции элементов, образующих термостойкие оксиды и гидроксиды (алюминия, молибдена и т. п.). Вторичная реакционная зона (3) предпочтительна для измерения атомной абсорбции элементов, не образующих термостойких оксидов (медь, се ребро, цинк, марганец и т. п.). Измерение в этой зоне характеризуется наибольшей стабильностью и наименьшими шумами.
Образование свободных атомов в пламени является следствием большой совокупности процессов, включая:
• получение аэрозоля из раствора анализируемой пробы;
• испарение растворителя из капелек аэрозоля;
• испарение твердых частичек аэрозоля и диссоциацию молекул на атомы;
• процессы возбуждения и ионизации атомов, а также образования новых соединений в результате реакций с радикалами, анионами, атомами кислорода и углерода, имеющимися в пламени.
Несмотря на простоту этого способа атомизации, он имеет ряд серьезных ограничений, обусловленных прочими реакциями в пламени и малой продолжительностью пребывания частиц в нем (10 -3 с). Кроме того, пламена не безопасны в работе и требуют расходов довольно больших объемов газообразных горючего и окислителя.
Более дешевыми, безопасными и эффективными во многих отношениях оказались электротермические атомизаторы,
Атомизация в электротермических атомизаторах
Очень простой в эксплуатации является тонкостенная графитовая печь, схема которой показана на рис. 2.7.
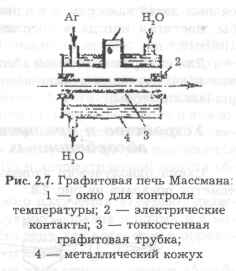
Анализируемую пробу в виде раствора дозируют микропипеткой в количестве 5-100 мкл через центральное отверстие на стенку холодной печи, концы которой закреплены внутри массивных графитовых контактов. Печь постоянно обдувается потоком аргона, что предохраняет ее от обгорания и способствует удалению испаренной пробы из атомизатора.
После высушивания проба испаряется до атомов, и атомный пар заполняет всю трубку.
Температура графитовой печи регулируется специальным электронным устройствам с программным управлением.
Обычно температурную программу по времени можно разделить на 3 этапа: высушивание пробы (испарение ра-створителя), озоление (пиролиз органических компонентов и удаление некоторых других компонентов матрицы), атомизация, т. е. собственно испарение и переход определяемого элемента в состояние атомного пара.
Каждому этапу соответствует своя оптимальная температура. Необходимость в такой ступенчатой температурной программе связана с тем, что на стадиях высушивания и озоления часто наблюдается другой сигнал абсорбции, обусловленный рассеянием зондирующего излучения дымом, частицами золы и т. д.
Атомизация пробы в графитовой печи в зависимости от физико-химических особенностей определяемых элементов и матриц пробы может происходить двумя путями:
• проба первоначально испаряется с нагретой поверхности атомизатора, а затем диссоциирует на элементы в газовой фазе;
• проба первоначально термически диссоциируется до соответствующих оксидов, которые затем восстанавливаются до металла, либо углеродом до твердофазной реакции на границе поверхностей:
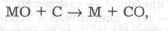
либо оксидом углерода:
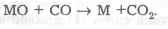
Метод атомной абсорбции с применением электротермического атомизатора обеспечивает рекордно низкие пределы обнаружения по многим элементам. Их численные значения
колеблются для разных элементов от десятых до десятитысячных долей нанограмма в одном миллиметре раствора пробы, достигая иногда в абсолютном выражении значения 10-12_10-4 г
Для измерения атомной абсорбции применяют однолучевые и двухлучевые атомно-абсорбционные спектрофотометры (анализаторы).
А́ТОМНО-АБСОРБЦИ́ ОННАЯ СПЕКТРОМ Е́ТРИ́Я (ААС), раздел оптич. спектроскопии, изучающий закономерности поглощения (абсорбции) оптич. излучения свободными атомами в газовой фазе. Возникновение ААС можно отнести к 1802, когда У. Волластон обнаружил тёмные линии в спектре излучения Солнца, позже (1814–15) объяснённые Й. Фраунгофером поглощением света в солнечной атмосфере. Широко используется в физике и астрофизике (определение коэф. диффузии атомов, темп-ры газа, химич. состава звёзд и др.), физич. химии (определение давления насыщенных паров, теплот испарения и др.). Наибольшее применение ААС нашла в аналитич. химии, составляя основу атомно-абсорбционного анализа (ААА) – метода количественного определения элементного состава вещества по спектрам поглощения атомов. В основе аналитич. приложений лежит избират. поглощение атомом оптич. излучения строго определённых длин волн. Как спектроаналитич. метод впервые реализован австрал. физиком А. Уолшем в 1955.
Абсорбционный спектральный анализ применяют для изучения спектров поглощения анализируемых веществ. Каждое вещество поглощает определенное количество света, обусловленное его природой или концентрацией. [1]
Абсорбционный спектральный анализ в ультрафиолетово видимой и инфракрасной областях спектра. Различают спектр фотометрический и фотоколориметрический методы. Спектроф тометрический метод анализа основан на измерении поглощен. Фотоколориметрический метод анализа основан на измерею светопоглощения или определения спектра поглощения в пр ] борах-фотоколориметрах в видимом участке спектра. [2]
Абсорбционный спектральный анализ подразделяют на спектрофотометрический и фотометрический методы. [3]
Абсорбционный спектральный анализ для обнаружения тория используют довольно редко. [5]
Абсорбционный спектральный анализ применяют для изучения спектров поглощения анализируемых веществ. Каждое вещество поглощает определенное количество света, обусловленное его природой или концентрацией. [7]
Абсорбционный спектральный анализ подразделяют на спек-трофотометрический и фотометрический методы. [8]
Абсорбционный спектральный анализ основан на способности атомов и молекул поглощать электромагнитное излучение. [9]
Абсорбционный спектральный анализ основан на изучении спектров поглощения анализируемых веществ. [10]
Абсорбционный спектральный анализ основан на наблюдении спектров поглощения анализируемых веществ. [11]
Абсорбционный спектральный анализ , основанный на идентификации отдельных полос и линий в спектрах поглощения атомов и молекул, позволяет получать данные о составе поглощающих систем, о тех атомах и группах атомов, которые входят в состав исследуемого вещества. [12]
Молекулярный абсорбционный спектральный анализ - фармакопейный метод и используется очень часто в качественном и количественном анализе многих лечебных препаратов - как субстанций, так и лекарственных форм. [13]
Схема проведения абсорбционного спектрального анализа ( см. рис. 1, б) отличается от уже рассмотренной схемы только в своей начальной части. Источником света служит нагретое твердое тело или другой источник сплошного излучения. Анализируемую пробу помещают между источником света и спектральным аппаратом. [14]
При проведении абсорбционного спектрального анализа излучение источника света, разложенное в спектр в монохроматоре, необходимо принять приемником, а затем зарегистрировать. Фотоэлектрическая регистрация спектров сводится к усилению и регистрации электрических сигналов, возникающих в термо - или фотоприемниках под действием падающего излучения. [15]
Читайте также:
- Запеканка из печени как в детском саду пошаговый рецепт
- Как вы думаете быть справедливым сложно обоснуйте свой ответ кратко для ученика 4 класса
- Какие меры предосторожности надо принимать чтобы не получить солнечный ожог кратко
- Как подбирают диаметр шканта и глубину отверстия под шкант ответы кратко
- Суворов как патриот россии кратко