
Синдром ангельмана кратко сообщение
Обновлено: 02.07.2024
Синдром Ангельмана (синдром счастливой марионетки) - этиология, клиника, диагностика
а) Патогенез. Синдром Ангельмана в большинстве случаев вызван делецией 15q11.2-12 хромосомы, сходной, но не идентичной выявляемой у детей с синдромом Прадера-Вилли (Magenis et al., 1987, Knoll et al., 1989) и наследуется по материнской линии (Knoll et al., 1989). Делеция включает ген бета-3-субъединицы ГАМК-рецептора (Saitoh et al., 1994). Среди 60-75% пациентов отмечаются делении или перестройки длинного плеча 15-й хромосомы, делеция всегда располагается на материнской хромосоме.
В небольшом количестве случаев выявляется дисомия отцовской 15-й хромосомы (Malcolm et al., 1991, Prasad и Wagstaff, 1997). Однако не менее чем у 15% пациентов выявляются нормальные хромосомы и отсутствуют признаки дисомии. В некоторых случаях такого рода возможно повторное рождение больных детей среди родственников (Clayton-Smith, 1992). Такие случаи могут быть связаны с доминантной мутацией гена UBE3A (Kishino et al., 1997) на 15q11-13 хромосоме, приводящей к возникновению фенотипа Ангельмана только при передаче по женской линии (Wagstaff et al., 1993).
На основании апраксической походки, стереотипиях движений рук и (в некоторых случаях) гипервентиляции возможна ошибочная постановка диагноза синдрома Ретта у девочек. Тем не менее, при раннем (до года) начале припадков, жизнерадостном настроении и дисморфизме вероятность неверной диагностики можно исключить.
в) Клинические проявления. Клинические проявления включают серьезную умственную отсталость с тяжелым нарушением речи, атаксию и заметные спастические нарушения движений верхних конечностей, а иногда и туловища; данные двигательные нарушения являются особым типом миоклонуса (Guerrini et al., 1996; Beckung et al., 2004). Немотивированные приступы смеха являются характерным, но не основным проявлением, в то время как жизнерадостное настроение является постоянным признаком (Williams и Frias, 1982).
У 86% больных детей отмечаются приступы, которые часто имеют повторный характер, но длятся недолго и чаще всего являются не тонико-клоническими припадками, а атипичными абсансами или тоническими или атоническими припадками (Dorries et al., 1988; Viani et al., 1995; Laan et al., 1997). На ЭЭГ часто встречаются характерные изменения в виде эпизодов медленных (3 Гц) волн, особенно в задних отделах, часто зазубренных и в отдельных случаях связанных с истинными пиками (Boyd et al, 1988).
На КТ и МРТ каких-либо специфичных изменений не выявляется, но возможно наличие небольшого расширения желудочков и/или околомозгового пространства (Dorries et al., 1988).
Способность к передвижению появляется поздно, часто после 5-6 лет; походка имеет аномальный характер с широко расставленными ногами и атаксическими и апраксическими признаками (Sugimoto et al., 1992).
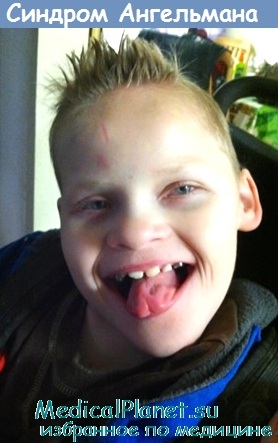
Дисморфический синдром характеризуется умеренной выраженностью и может быть незаметен в детском возрасте. Прогнатизм обычно развивается только в возрасте нескольких лет.
Только недавно были опубликованы результаты исследований, подтверждающих мнение о специфическом поведенческом фенотипе данного синдрома, и еще слишком рано делать предположения в данном направлении. Результаты одного из исследований взрослых и молодых пациентов, страдающих синдромом Ангельмана (Clayton-Smith, 1993), свидетельствуют о том, что гиперактивность, часто наиболее выраженная в раннем детском возрасте, в позднем детском возрасте обычно сменяется более контролируемым поведением.
Результаты большинства исследований в настоящее время позволяют предположить, что мутизм может быть постоянным проявлением, но некоторые пациенты могут освоить язык жестов. В одном из центров наблюдался мальчик с сочетанием синдрома Ангельмана и аутизма. У многих детей с синдромом Ангельмана отмечаются заметные аутистические проявления, такие как упрямство, настаивание на одном и том же и увлеченность водой, и в то же время они кажутся счастливыми и общительными, так как им нравится контактировать с другими людьми. Тем не менее, контакт осуществим лишь при выполнении их собственных условий.
г) Исход. Прогноз неблагоприятный, отмечается серьезная умственная отсталость. Овладение коммуникативным языком невозможно (Beckung et al., 2004).

Синдром Ангельмана (СА) – генетическое заболевание, которое вызвано хромосомными нарушениями. Оно имеет несколько названий, отражающих проявления болезни: синдром счастливой куклы, а также марионетки или Петрушки
Синдром Ангельмана у детей становится заметным в первые годы жизни. При рождении ребенок с генетическим дефектом кажется абсолютно здоровым. Первые симптомы проявляются к годовалому возрасту.
При подобной патологии наблюдаются неврологические расстройства, отставание в психическом развитии, возникают двигательные трудности, проявляются симптомы эпилепсии.
Синдром счастливого Петрушки характеризуется следующими признаками: радостное лицо, приступы беспричинного смеха, марионеточная походка. Такие дети доброжелательны, эмоциональны, любят общение, часто смеются, всегда улыбаются.

Синдром Ангельмана: клинические симптомы
Впервые болезнь описал английский педиатр Г. Ангельман. Ее признаки бывает трудно определить до года. Явно выраженные симптомы проявляются с двухлетнего возраста. К этому времени уже заметны клинические проявления:
1. задержка психического развития – снижение когнитивных функций, умственная отсталость
2. речевые нарушения – ограниченная речь или ее отсутствие
3. психомоторные расстройства – хаотичные движения рук, тремор конечностей, атаксия (проблемы с координацией), специфическая походка
4. нарушения физического развития – черепно-лицевые аномалии: микроцефалия (объем черепа меньше нормы), редкие зубы, слюнотечение, уплощенный затылок, выступающая нижняя челюсть, открытый рот, высунутый язык. часто наблюдается сколиоз, мышечная дистония, косоглазие
5. эпилептическая активность, приступы судорог
6. особенности поведения (гиперактивность, смех без повода)
7. беспокойный сон.
Синдром Ангельмана у взрослых может быть менее выражен. С возрастом клинические проявления ослабевают – снижается гиперактивность, улучшается сон, уменьшается частота судорожных приступов. У них часто проявляются ожирение и сколиоз.
Внешние признаки при легкой форме болезни могут быть незаметными. Многие взрослые выглядят значительно моложе своих лет. Из-за выраженной задержки в развитии они в душе остаются детьми – могут быть импульсивными, чрезмерно обидчивыми, а иногда агрессивными.
Синдром Ангельмана: основные причины заболевания
Различают несколько вариантов генетической патологии при СА. Чаще всего причина заболевания – генная мутация определенного участка 15 хромосомы матери. Аномалия обусловлена делецией – потерей хромосомного сегмента.
Могут быть и другие причины:
• трисомия – наличие лишней хромосомы
• микроделеция – отсутствие одного гена
• инверсия – нарушение генного порядка из-за разворота отдельного участка хромосомы
• транслокация – перенос части одной хромосомы к другой.
![]()
Диагностика

Многие пациенты наблюдаются у специалистов с эпилепсией, речевой задержкой, различными нарушениями поведения, не зная о своем заболевании. Для постановки диагноза необходима консультация невролога, психиатра, врача-генетика.
Выявлять аномалии, которые приводят к возникновению синдрома Ангельмана, позволяют исследования: генетический анализ, МРТ, ЭЭГ, УЗИ. Самыми надежными считаются генетические исследования. На основании их результатов при характерном проявлении симптомов заболевания можно установить точный диагноз.
Диагностировать АС можно еще до рождения ребенка во время беременности. Для этого применяют два вида исследований:
1. неинвазивные – скрининги, включающие комплексное исследование по выявлению у плода хромосомных аномалий. Достоверность результатов не всегда подтверждается
2. инвазивные – высокоточные методы, их назначают женщинам старше 35 лет для подтверждения результатов, полученных другими методами. Они небезопасные, проводятся только по показаниям, учитывая высокий риск осложнений.
Лечение и профилактика
Влиять на генетический дефект невозможно, нарушения происходят на хромосомном уровне, поэтому больным назначают симптоматическое лечение и психологическую коррекцию.
Медикаментозная терапия может применяться при эпилептических приступах. Для смягчения симптомов заболевания и улучшения сна могут использоваться успокаивающие препараты.
Разрабатываются индивидуальные программы для каждого пациента – ЛФК, логопедические занятия, лечебный массаж, поведенческая терапия. Они дают возможность улучшить качество жизни человека. Также привлекают логопедов, дефектологов, специалистов по невербальным способам общения и поведенческой терапии.
В профилактических целях парам, планирующим зачатие ребенка, рекомендуется консультация генетика.
Прогноз
Тяжесть болезни может быть разной. Прогноз зависит от степени повреждения материнской 15 хромосомы. Некоторые пациенты могут себя обслуживать и общаться, другие – не способны передвигаться и говорить.
Проведение физиотерапии для развития точности движений, коррекция коммуникационных навыков улучшают прогноз. Но в любом случае больные не смогут стать абсолютно самостоятельными, им требуется помощь на протяжении всей жизни.
Синдром Ангельмана – это генетическое заболевание, сопровождающееся отставанием в психическом развитии. Болезнь сочетается с припадками, с немотивированным смехом и хаотическими движениями рук. Подобные проявления у больных людей способствовали тому, что синдром Ангельмана часто называют синдромом Петрушки или синдромом смеющейся куклы, либо счастливой куклы.
Эпоним данный синдром получил от врача-педиатра Б. Г. Ангельмана, который впервые описал ее в 1965 году. Согласно статистике, синдром Ангельмана встречается у 1 ребенка из 10-20 тысяч.
Симптомы синдрома Ангельмана
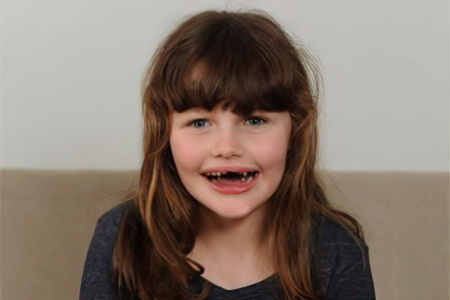
Симптомы синдрома Ангельмана многообразны.
У некоторых больных они проявляются чаще, у других реже, но в целом клиническая картина выявляется следующая:
Внешние признаки: окружность головы больного меньше средних значений, зубы расположены редко, затылок уплощен. Разрез рта у больного широкий, из него виден язык, который часто высунут наружу, подбородок при этом выдается вперед. Иногда диагностируется косоглазие.
Сразу после рождения начинаются проблемы с питанием. Дети плохо кушают, вес набирают с трудом.
Всегда присутствует ЗПР, в раннем возрасте дети поздно начинают садиться и ходить.
Ребенок хорошо понимает, что до него пытаются донести, но высказать свою мысль может с трудом. Страдает речевое развитие.
Гиперактивность – характерная черта таких детей. Движения конечностями при этом хаотичны, заметен мелкий тремор.
Страдает внимание, в первую очередь такой процесс, как концентрация. Это приводит к проблемам в обучении.
Нередки эпилептические припадки.
Улыбка ребенка беспричинная, смех немотивирован.
Характерна для больных с синдромом Ангельмана марионеточная походка, то есть ноги во время движения сгибаются в коленных суставах минимально.
Всегда имеются проблемы со сном.
Больные плохо переносят высокие температуры, хорошо чувствуют себя в воде.
Если рассматривать частоту встречаемости симптомов, то у больных всегда имеются проблемы с психическим и физическим развитием, страдает речь, поведение, моторика, концентрация внимания. В 80% случаев у детей случаются приступы эпилепсии.
Реже наблюдаются судороги, деформация черепа. Совсем нечасто выявляются проблемы с питанием, косоглазие, дрожащий язык.
Случается, что по мере взросления больного, симптоматика несколько меняется. При этом все люди с синдромом Ангельмана выглядят младшего своего возраста. Некоторые из обзаводятся семьями, однако риск рождения ребенка с аналогичной патологии высока.
С возрастом у больных прогрессирует сколиоз, имеется склонность к ожирению.
Причины синдрома Ангельмана
Причины синдрома Ангельмана кроются в генетических аномалиях, так как в 15 хромосоме у больных людей отсутствует часть генов. Эти хромосомные нарушения объясняются явлением делеции, либо мутациями.
Синдром Ангельмана развивается в том случае, когда нарушения происходят в материнской хромосоме, если изменяется отцовская хромосома, то у больного диагностируют синдром Прадера-Вилли.
Ведущей причиной патологии называют нарушения в делении 15 хромосомы, которые влекут за собой аномалии в создании копий материнских генов. Реже к развитию болезни приводит мутация отцовских генов, отцовская трисомия и дисомия.
Так, здоровые люди получают от обоих родителей по 1 копии 15 хромосомы, когда ребенок получает от одного из родителей мутированную копию (чаще всего от матери), то у него развивается данный синдром.
Диагностика синдрома Ангельмана
Диагностика синдрома Ангельмана выстраивается на основе проведения генетического исследования 15 хромосомы. Этот анализ показан детям с гипотонусом, с нарушениями психического и физического развития.
Основанием для генетического анализа служат симптомы, характерные для синдрома Ангельмана. Стоит отметить, что существует отдельная группа людей, у которых все анализы остаются в норме, а признаки болезни присутствуют.
Лечение синдрома Ангельмана

Лечение синдрома Ангельмана сводится к выполнению определенных мероприятий, которые позволяют улучшить качество жизни больного. Медикаментозная терапия является бессильной, так как нарушения происходят на хромосомном уровне. Поэтому в детстве показан лечебный массаж для коррекции гипотонуса, а также выполнение укрепляющих физиопроцедур.
По мере взросления ребенка, ему необходима помощь логопеда. Регулярные занятия позволяют добиться более четкого произношения и артикуляции.
При проблемах со сном, рекомендуют к приему снотворные препараты. Так, пациентам до ночного отдыха (за 30 минут), рекомендуют прием мелатонина в дозировке 0,3 г. Эпилептические припадки, тремор конечностей и судороги с успехом купируются противосудорожными средствами.
Легкие слабительные препараты назначают при проблемах со стулом.
Врачи в США проводят терапию синдрома Ангельмана с помощью гормона Секретин, который используется для лечения детей с аутизмом. Препарат вводится внутривенно и способствует снижению симптомов заболевания, нормализуя поведение и улучшая коммуникативные навыки.
При этом, люди с синдромом Ангельмана не ущербны: они очень общительны и дружелюбны, хорошо ладят с детьми, всегда в хорошем настроении. Недостаточность коммуникативных навыков возможно заместить невербальными средствами общения.
Что касается прогноза, то высок риск того, что второй ребенок в семье будет также страдать синдрома Ангельмана, поэтому, чтобы этот риск минимизировать, необходима консультация у генетика. Дело в том, что если мутация не унаследованная, то вероятность появления второго больного ребенка сводится всего к 1%.
Прогноз на жизнь в целом благоприятный, хотя зависит от степени поражения хромосомы. Так, некоторые больные полноценно осваивают навыки самообслуживания, способны общаться с окружающими людьми. Иные пациенты, напротив, не способны контролировать даже собственный стул, не в состоянии самостоятельно передвигаться и разговаривать.
Во взрослом возрасте проблему представляет сколиоз, а также ожирение со всеми последствиями этого состояния.

Автор статьи: Мочалов Павел Александрович | д. м. н. терапевт
Образование: Московский медицинский институт им. И. М. Сеченова, специальность - "Лечебное дело" в 1991 году, в 1993 году "Профессиональные болезни", в 1996 году "Терапия".
Наши авторы
Харьковский специализированный медико-генетический центр (ХСМГЦ) занимается исследованиями в генетике и эпигенетике, диагностикой и лечением аутизма, дефицита внимания, слуховой нейропатии, муковисцидоза, нарушений метаболизма, обмена аминокислот, а также других редких генетических (орфанных) заболеваний.
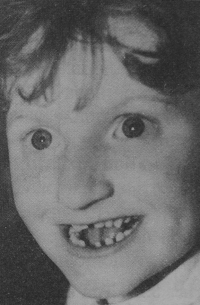
Что такое Синдром Ангельмана?
Причины
Синдром Ангельмана возникает вследствие потери нормальных материнских копий генов в определенной области 15 хромосомы. Чаще всего это происходит путем делеции сегмента этой хромосомы. Другими причинами возникновения заболевания могут быть одноотцовская дисомия, транслокация или мутация одного гена в этой области. Обычно, здоровый человек получает две копии 15 хромосомы, одну от матери, другую от отца. Однако, область этой хромосомы, которая является очень важной для синдрома Ангельмана, экспрессия материнских и отцовских копий генов очень отличается. Это связано с гендерным эпигенетических импринтингом; биохимическим механизмом метилирования ДНК. У здорового человека, экспрессия материнской аллели - сильнее, тогда как отцовская аллель почти не проявляется. Если же материнские копии генов - удалены или мутированы, то это вызывает появление синдрома Ангельмана (если вследствие аналогичных механизмов потеряна отцовская копия, то это вызывает синдром Прадера-Вилли).
Диагноз синдрома Ангельмана обычно ставится, когда ребенку исполняется
3 -7 лет, и признаки болезни становятся явно выраженными. Заболевание это нельзя обнаружить у новорожденных детей или в первый год жизни, так как его симптомы в таком раннем возрасте — неспецифические.
В поздней диагностике виновата еще и малая осведомленность об этой патологии врачей семейной медицины и родителей. В настоящее время этот синдром диагностируется все раньше, благодаря большей информации об этом заболевании, которая в развитых странах распространяется общественными объединениями родителей больных детей.
Начальные исследования
Кариотип 46 XX или XY, 15р−. Обычно синдром вызывается спонтанным хромосомным дефектом, когда отсутствует большая смежная область из 3—4 миллионов пар оснований ДНК в области q11—q13 15-й хромосомы.
Частота встречаемости, по разным данным, — 1 : 10 000—20 000 живорожденных младенцев.
Начальные исследования, которые проводятся с помощью мышей, у которых отсутствует экспрессия материнской копии гена UBE3A, обнаружили серьезные отклонения в деятельности гиппокампа, которые нарушают процесс формирования памяти. В частности, наблюдаются отклонения в парадигмах обучения, особенно в тех процессах, которые зависят от деятельности гиппокампа - кондиционирование страхом.
Клинические проявления
- тяжелая функциональная задержка развития;
- нарушение речевых функций (полное отсутствие или минимальное использование языка); невербальные навыки развиты лучше, чем вербальные;
- расстройство моторных функций (нарушения движения, равновесия), как правило, атаксия и (или) дрожание конечностей;
- поведенческие отклонения: сочетание частого беспричинного смеха (улыбки), счастливого состояния, легкой возбудимости, частого хлопанья в ладоши, гипермоторного поведения, низкий уровень концентрации внимания.
Для синдрома Ангельмана характерны:
- В 75 % проблемы с питанием, особенно с грудным вскармливанием, такие младенцы плохо набирают вес;
- дети больше понимают, чем могут сказать или выразить;
- дефицит внимания и гиперактивность;
- сложности с обучением; микроцефалия;
- эпилепсия (80 % случаев), нарушения выявляются также при электроэнцефалографии; считается, что у детей с синдромом Ангельмана
- наблюдается вторичная (симптоматическая) эпилепсия;
- необычные движения (мелкий тремор, хаотические движения конечностей);
- ходьба на негнущихся ногах — из-за этой особенности детей с этим синдромом иногда сравнивали с марионетками;
- размер головы меньше среднего, нередко с уплощением затылка;
- иногда характерные черты лица — широкий рот, редко расположенные зубы, выдающийся вперед подбородок, высунутый наружу язык;
- нарушения сна;
- страбизм (косоглазие) в 40 % случаев;
- сколиоз (искривление позвоночника) в 10 % случаев;
- повышенная чувствительность к высокой температуре;
- чувствуют себя комфортнее в воде.
Связанные характеристики (характерные для 20 - 80% больных):
- косоглазие;
- гипопигментация кожи и глаз;
- нарушение контроля над движениями языка, трудности при сосании и глотании;
- гиперактивные сухожильные рефлексы;
- проблемы с питанием в раннем детстве;
- подняты, согнуты во время шествия руки;
- выдвинута нижняя челюсть;
- повышенная чувствительность к теплу;
- широкий рот, широкий интервал между зубами;
- нарушение сна; - частые слюнотечения, высунутый язык;
- постоянное желание пить;
- усиленные жевательные движения;
- плоский затылок;
- гладкие ладони.
Болезнь Ангельмана в социуме
Больные СА, как правило, люди, которые имеют счастливый вид, они любят общаться с людьми и играть в разные игры. Однако, установление связи с другими людьми, общение с ними может сначала быть несколько трудным, но, поскольку дети с СА постепенно развиваются, у них возникает определенный характер, появляются разные способности и их начинают лучше понимать другие люди. В частности, люди с синдромом Ангельмана очень часто применяют невербальные средства общения, для того, что бы компенсировать ограниченные возможности речи. Часто, врачи отмечают, что их понимание развито гораздо больше, чем коммуникационные способности. Большинство больных, применяют, как правило, лишь 10-15 слов, если вообще разговаривает.
Существует ли лечение синдрома Ангельмана?
Синдром Ангельмана является врожденной генетической аномалией; в настоящее время специфические способы его лечения не разработаны.
Однако некоторые лечебные мероприятия повышают качество жизни людей с синдромом.
В частности, младенцы с гипотонусом должны получать массаж и другие виды специальной терапии (физиотерапии).
Рекомендуется использование специальных методик развития ребенка, занятия с логопедом и дефектологом.
Нарушения сна корректируются назначением легких снотворных. Нарушения стула регулируются назначением легких слабительных.
Приступы лечатся так же, как эпилепсия.
Прогноз
Тяжесть симптомов, возникающих при синдроме Ангельмана, отличаются в каждом конкретном случае. Некоторые люди, с легкой формой синдрома Ангельмана, имеют большой словарный запас и высокую степень самоконтроля. Однако ходьба и использования простых языковых знаков может пострадать значительно сильнее. Раннее начало и постоянное проведение физиотерапии (которая направлена на развитие точных движений) и коммуникационных свойств (языки), как полагают, исследователи, значительно улучшает прогноз (когнитивного и речевого развития) для больных СА.
Подготовил студент 5 курса 1 группы
лечебного факультета
Миоков С.А.
Читайте также: