
Ретинобластома у детей реферат
Обновлено: 05.07.2024
Ретинобластома — редкая злокачественная опухоль глаз у детей с частотой 1 на 18000 живорожденных. Поздняя диагностика приводит к смертности в 70% случаев; при доступности оптимальной терапии более 95% детей вылечиваются. Эффективным подходом в лечении ретинобластомы является мультидисциплинарный подход с участием как клиницистов (офтальмологи, детские онкологи и лучевые терапевты, медицинские сестры, генетики), так и специалистов в области получения изображений, детских (игровых) специалистов и родителей. Национальные руководства могут способствовать разработке стандартов и созданию условий для проверок, исследований и клинических испытаний для постоянного развития системы оказания помощи и улучшения результатов лечения.
Опухоли происходят из эмбриональных клеток сетчатки, поэтому в большинстве случаев развиваются в возрасте до четырех лет. Первичное лечение включает выполнение энуклеации и проведение химиотерапии в комбинации с лазерокоагуляцией и криотерапией. Пациенты с системной мутацией гена-супрессора опухолевого роста RB1 находятся в повышенной зоне пожизненного риска развития других злокачественных опухолей, риск повышается при радиационном облучении. Более того, лучевая терапия в настоящее время не является лечением первого выбора с целью сохранения глаза, поэтому скрининговое обследование на экстраокулярную и трехстороннюю (трилатеральную) ретинобластому предпочтительно проводить при помощи МРТ и ультразвукового исследования, а не КТ.
Исследование ретинобластомы являлось основополагающим в понимании рака в целом. Исследования ретинобластомы обнаружили, что наследственные и ненаследственные опухоли развиваются при потере обеих аллелей гена-супрессора опухолевого роста RB1. На основании данных клинических исследований ретинобластомы было предположено существование специфического гена, подавляющего развитие злокачественной опухоли.
Ген RB1 был первым клонируемым геном-супрессором опухолевого роста, и было обнаружено, что он играет ведущую роль в развитии многих видов злокачественных опухолей.
Патогенез ретинобластомы
а) Наследственная и ненаследственная ретинобластома. У всех детей с двусторонней (билатеральной) ретинобластомой в одной из хромосом (13) имеется мутация гена RB1, предрасполагающая к развитию опухолей сетчатки в младенческом возрасте и других опухолей в течение жизни. Несмотря на то, что в 90% случаев отсутствует предшествующий семейный анамнез ретинобластомы и у впервые заболевших в этих семьях имеется новая мутация зародышевой линии, у 50% их потомства мутантный ген RB1 передастся и разовьется опухоль. У большинства детей без семейного анамнеза ретинобластомы только в одном глазу имеются нормальные врожденные аллели RB1, но на глазу с развившейся опухолью происходит потеря обеих функциональных аллелей, напоминая картину наследственной опухоли.
У 15% лиц с развившейся односторонней ретинобластомой были врожденные мутации аллели RB1, которые могут передаться их потомству.
Молекулярная и клиническая генетика являются неотъемлемой частью лечения во всех семьях, пораженных ретинобластомой.
б) Потеря обеих аллелей гена RB1 вызывает развитие ретинобластомы. Наблюдение, согласно которому у детей с двусторонней ретинобластомой заболевание диагностируется в более раннем возрасте, чем с ненаследственной, привело к теории Нудсона, утверждающей, что для начала развития ретинобластомы необходимы две мутации. На основе этого анализа предположено, что вторая одиночная мутация в развивающейся клетке сетчатки, с уже имеющейся предрасполагающей врожденной мутацией, вызывает развитие опухоли (наследственная ретинобластома), но при ненаследственной односторонней форме ретинобластомы в одной развивающейся клетке сетчатки имеются обе мутантные аллели.

(А) Семейное дерево: матери проводили лечение двусторонней ретинобластомы энуклеацией глаза и дистанционной лучевой терапией другого глаза.
Через 42 года в зоне облучения у нее развились метастазы гемангиосаркомы (красная).
Роды обоих детей были проведены на 36 неделе гестации для облегчения раннего лечения двусторонних опухолей.
Мать и оба ребенка являются носителями зародышевой мутации RB1, что приводит к отсутствию pRB при потере нормальной аллели RB1 (М2) в развивающейся клетке сетчатки, вызывая развития опухоли.
(Б) Изображения камеры RetCam®: в три месяца на правом глазу мальчика до лечения видны две опухоли (группа А по IIRC, опухоль более чем в 1,5 мм от диска зрительного нерва);
стабильное состояние правого глаза в четыре года после лазерокоагуляции и двух курсов КЭВ (карбопластин, этопозид и винкристин) химиотерапии с циклоспорином А и еще большим числом курсов лазерокоагуляции.
(В) Изображения камеры RetCam®: до лечения, левый глаз девочки в два месяца (группа В по IIRC, опухоль менее чем в 3 мм от фовеа);
рубцы после лазерокоагуляции и новая опухоль над зрительным нервом в четыре месяца;
рецидив в области исходного рубца с распространением в сторону фовеа с васкуляризацией опухоли по данным флюоресцентной ангиогарфии;
плоские рубцы в 2,5 года после проведения лазерокоагуляции, двух курсов КЭВ химиотерапии с циклоспорином А для контролирования,
угрожающих зрению, повторного проведения химиотерапии и в большем объеме лазерокоагуляции (иллюстрации Leslie MacKeen, Cynthia VandenHoven и Carmelina Trimboli.)
Мутация зародышевой линии RB1 приводит к 40000-кратному относительному риску (ОР) развития ретинобластомы, 500-кратному ОР развития саркомы, который возрастает до 2000-кратного при применении лучевой терапии, но не повышается при лейкозе. Хотя pRB является ключевым во всех делящихся клетках, его роль в развитии клеток крайне тканеспецифична. Часть развивающихся клеток сетчатки может зависеть исключительно от pRB для окончательной дифференцировки во взрослую, функционирующую сетчатку. Потеря pRB способствует геномным изменениям и нестабильности, приводящей к последующим мутациям в онкогенах и других генах-супрессорах опухолей, что приводит к развитию опухоли сетчатки.
д) Другие манифестации мутантных аллелей гена RB1. Мутация RB1 также предрасполагает к доброкачественным опухолям сетчатки, ретиноме, эктопической интракраниальной ретинобластоме (трилатеральная (трехсторонняя) ретинобластома), и второй экстраокулярной злокачественной опухоли.
е) Ретинома. Ретинома — доброкачественный вариант мутации гена RB1, Эту непрогрессирующую патологию характеризуют три признака: проминирующая серая масса сетчатки, кальцинаты и окружающие их пролиферирующий пигментный эпителий сетчатки (ПЭС) и пигментация. Эти три признака также видны и после проведения лучевой терапии по поводу ретинобластомы. Ретинома, если она выявлена в детстве, что бывает очень редко, бессимптомная опухоль без озлокачествления. Описаны единичные случаи, когда ретинома прогрессировала в активную ретинобластому. Однако как правило, ретинома располагается под активной ретинобластомой и может быть обнаружена при патогистологическом исследовании энуклеированного (удаленного) глаза. Отличительной особенностью является образование цветочноподобных клеток и отсутствие маркеров пролиферации.
При ретиноме обе аллели RB1 мутантные и определяется геномная нестабильность, которая прогрессирует в степени и числе генов, вовлеченных в смежной, быстро пролиферирующей ретинобластоме. Обнаружение ретиномы при осмотре сетчатки у родственника пациента с ретинобластомой указывает, что они несут мутантную аллель RB1.
ж) Эктопическая внутричерепная (трилатеральная, трехсторонняя) ретинобластома. Трехсторонняя (трилатеральная) ретинобластома это среднемозговая опухоль или первичная опухоль пинеальной области, связанная с наследственной ретинобластомой, но не вследствие метастазирования. Опухоль относится к нейробластомам и напоминает низкодифференцированную ретинобластому. Пинеальные опухоли развиваются у 5% детей с мутациями гена RB1, но не следует их путать с кистами шишковидной железы, встречающимися у 2% всех детей и не требующими лечения. У больных детей может быть повышенное внутричерепное давление при отсутствии признаков новообразования в пинеальной или параселлярной областях по данным МРТ. Опухоль шишковидной железы можно выявить при помощи МРТ при рутинном скрининге на интракраниальные опухоли на стадии, когда еще можно провести лечение.
з) Множественные разные злокачественные опухоли. У людей с мутантным геном RB1 повышенный риск развития второй экстраокулярной злокачественной опухоли, которая может развиваться внутри или вне зоны облучения. Облучение, особенно у младенцев до года жизни, повышает риск развития сарком и других злокачественных опухолей внутри зоны облучения. Наиболее характерной второй первичной опухолью у лиц с мутациями гена RB1 является остеосаркома, но имеются публикации и о множестве других новообразований. Эти индуцированные облучением опухоли крайне сложны в лечении, раньше дети с мутациями в гене RB1 чаще умирали от второй опухоли, чем от неконтролируемой ретинобластомы. В настоящее время у детей с ретинобластомой облучение ограничивают, чтобы сохранить оставшийся глаз.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021



Современные представления о ретинобластоме
Реферат
Представлен обзор отечественной литературы, который отражает современные представления о наиболее частой злокачественной опухоли глазного яблока среди детей. Освещены основные патогенетические механизмы образования ретинобластомы, факторы риска, способствующие формированию данной патологии. Отражены первые признаки заболевания и описаны клинические проявления данной опухоли. Кратко рассмотрены вопросы терапии ретинобластомы. Разобраны два основных метода лечения, таких как химиотерапия и хирургические методы. Рассмотрена программа реабилитации, указаны благотворительные организации и медицинские учреждения, в которых дети могут получить материальную, психологическую, педагогическую и медицинскую помощь, а также диспансерное наблюдение детей с ретинобластомой и меры профилактики для предотвращения рождения ребенка с данной патологией.
Ключевые слова: ретинобластома, нейроэктодермальная опухоль сетчатки, офтальмоонкология
Modern ideas about retinoblastoma
Borisova Natal'ia Aleksandrovna, D'yachkova Tat'iana Aleksandrovna
Northern state medical university, Arkhangelsk, Russia
Students of the 6th course of pediatric faculty
Key words: retinoblastoma, neuroectodermal tumor of the retina, ophthalmooncology
В последние десятилетия отмечается рост заболеваемости ретинобластомой (РБ), что связывают с ухудшением экологической обстановки, повышением радиационного фона Земли и совершенствованием диагностических возможностей медицинской науки. РБ является одной из наиболее частых детских злокачественных опухолей глазного яблока, встречающаяся во всех возрастных группах детского населения [7], приводящая к инвалидизации и необратимой слепоте, несмотря на современные методы диагностики.
В последние десятилетия отмечен рост заболеваемости в два раза. Если в 80-е годы прошлого столетия опухоль диагностировали у 1 из 34000 новорожденных, то в настоящее время - у 1: 15000-20000 новорожденных [3].
РБ – это злокачественная нейроэктодермальная опухоль оптической части сетчатки (внутренней оболочки глаза), чаще поражающая детей в возрасте от 0 до 9 лет [1]. Различают две формы: монокулярную (поражен один глаз) и бинокулярную (поражены оба глаза). Данная опухоль является генетически детерминированной (обусловленной) и формируется в эмбриональный период. В первые месяцы беременности под воздействием внешних факторов возникают генные мутации в половых клетках, что в дальнейшем приводит к тому, что клетки сетчатки начинают аномально расти и делиться, в результате чего образуется опухоль по типу бинокулярной формы. Если в семье нет родственников с диагнозом РБ глаза, то она является первичным заболеванием и, как правило, бывает монокулярной [4]. Причиной развития данной формы могут служить вредные привычки родителей, неблагоприятные условия труда и экологическая обстановка, дефицит пищевых нутриентов, перенесенные инфекционные заболевания, бесконтрольный прием лекарственных препаратов и др.
Основными задачами лечения являются сохранение жизни детей, сохранение органа зрения и его функций, профилактика рецидивов и метастазирования, а также реабилитация детей с утратой органа зрения [3].
Лечение РБ проводится в специализированных офтальмологических или онкологических центрах при участии врача - офтальмоонколога, онколога, радиолога, химиотерапевта. Для лечения применяются медикаментозные (химиотерапия), хирургические (ликвидационные, органосохраняющие) и лучевые методы. При своевременном и адекватном лечении выживаемость при монокулярных формах РБ составляет 99%, при бинокулярных – 95% [7].
Приоритетными задачами лечения РБ являются сохранение жизни ребенка, сохранение органа зрения и его функций, профилактика рецидивов и метастазирования [3]. Терапия РБ комбинированная. В ее состав, в настоящее время, входят химиотерапия и хирургические методы лечения. Химиотерапия (ХТ) используется как системная, так и локальная, которая в свою очередь подразделяется, в зависимости от пути введения, на суперселективную интраартериальную химиотерапию (СИАХТ) и интравитреальную химиотерапию (ИВХТ). Для этого применяются цитостатики: винкристин, этопозид, карбоплатин, циклофосфамид и др. Помимо химиотерапии, применяются хирургические методы лечения, которые подразделяются на ликвидационные, лучевые и локальные органосохраняющие методы лечения. К ликвидационным методам относятся энуклеация (удаление глазного яблока) и экзентерация (удаление содержимого глазницы). К локальным методам относятся лазеркоагуляция, транспупиллярная термотерапия и криодеструкция. Лучевые методы подразделяются на брахитерапию и наружное облучение глаза и орбиты. Выбор тактики лечения должен проводиться в зависимости от возраста пациента, стадии заболевания, размера и локализации опухоли, характера роста опухоли, одно- и двусторонности поражения, генетической предрасположенности [3].
Перед государством достаточно остро стоят вопросы о социальной и медицинской реабилитации детей с РБ. Дети нуждаются в данных видах реабилитации, направленных на повышение качества жизни ребенка и его семьи, а также в психологической помощи в развитии ребенка как личности и адаптации в обществе – посещение дошкольных и школьных учреждений.
Отсутствие глазного яблока, косметические дефекты, потеря зрения приводят к психологическому дискомфорту личности в обществе, высокому уровню инвалидности и низкому качеству жизни ребенка и его семьи. Дети нуждаются в социальной и медицинской реабилитации, а также в психологической помощи в развитии ребенка как личности и адаптации в обществе – посещение дошкольных и школьных учреждений. В связи с этим разработка определенных алгоритмов диагностических мероприятий, позволяющих вовремя выявить опухоль, и лечебных манипуляций, направленных на сохранение жизни ребенка, глаза и его функций, является важной задачей офтальмоонкологии. Обязательной частью реабилитации детей с анофтальмом является глазное протезирование [6]. Детей приучают к ношению протеза, который меняют по мере роста ребенка.
Финансовую, реабилитационную и социальную помощь активно оказывают различные фонды помощи детям с онкологическими заболеваниями: “Русфонд”, “Подари жизь”, “Настенька”, “Жизнь” и т.д., а также в этом принимают участие группы в социальных сетях, форумы.
Онкологическим больным с РБ показаны многие методы курортной терапии: климатотерапия, ландшафтотерапия, аэроионотерапия, терренкур, питьевое лечение минеральными водами, лечебная физкультура в залах и на природе, занятия в водоемах и бассейнах, скандинавская ходьба, иппотерапия, диетотерапия, которые в комплексе с необходимым медикаментозном лечением способствуют улучшению общего состояния больных [2]. Благодаря вышеперечисленным методикам больные перестают ощущать свои соматические нарушения и выходят из тяжелой стрессовой ситуации, связанной с его заболеванием и последствиями лечения.
Психологическая реабилитация играет огромную роль для восстановления здоровья ребенка, перенесшего онкологическое заболевание. Длительное и сложное лечение, изоляция от общества, тревога по поводу болезни, переживания из-за изменения внешности влияют на психологическое состояние детей. Целью программы является коррекция и профилактика психологических нарушений с рекомендациями для дальнейшего восстановления и повышения качества жизни.
Дети с данной патологией состоят на диспансерном учете у офтальмоонколога до 18 лет, затем передаются под наблюдение во взрослую сеть. Офтальмоонколог проводит осмотры 1 раз в квартал в течение первого года после лечения, затем один раз в полгода в течение 2-3 лет, затем один раз в год после 3 лет заболевания. Под диспансерным наблюдением также должны находиться дети раннего возраста (до 3 лет жизни), родившиеся в семьях, где имеются больные с РБ. Дети с любой формой РБ с диспансерного учета не снимаются [1].
Женщинам до планируемой беременности и во время нее рекомендуется вести здоровый образ жизни (сбалансированное питание, богатое витаминами и минералами, адекватные физические нагрузки, полноценный сон), тщательно следить за состоянием здоровья (предупреждать возникновение инфекционных заболеваний), полностью отказаться от различного рода зависимости (курение, алкоголь, наркотики), ограничить употребление лекарственных препаратов (антибиотики, некоторые анальгетики и др.), по - возможности избегать рентгенологических исследований, в т.ч. флюорографию, определяя ожидаемую пользу для матери и потенциальный риск для плода.
При решении создания семьи и детей обязательная генетическая консультация с проведением пренатальной диагностики. При семейной форме РБ для рождения здорового ребенка возможно использование процедуры экстракорпорального оплодотворения. По мнению ученых НИИ гематологии и онкологии, проведенные молекулярно-генетические исследования доказывают, что генетическое обследование детей и их родителей должно проводиться во всех случаях выявления РБ [1]. В настоящее время только это является залогом успеха рождения здорового ребенка.
Таким образом, при своевременной ранней диагностике РБ и адекватном своевременном лечении с помощью различных органосохраняющих методик и дальнейшей реабилитацией возможно сохранение органа зрения и его зрительной функции.
Список литературы
- Горовцова О.В., Иванова Н.В., Ушакова Т.Л. и др. Клинические рекомендации по диагностике и лечению ретинобластомы у детей и подростков /под редакцией Полякова В.Г. // НИИ детской онкологии и гематологии.- 2015. - 47с.
- Дыбов С.П. Ретинобластома: лечение, диагностика, реабилитация. - Самара.- 2011.-126 с.
- Саакян С.В, Катаргина Л.А. и др., Федеральные клинические рекомендации по диагностике, мониторингу и лечению детей с ретинобластомой // Ассоциация врачей-офтальмологов.- 2014. -30 с.
- Милованова, В.М. Панормова, Н.В. Вопросы офтальмогенетики. - Москва.-2010.- 315 с.
- Ушакова Т.Л. Современные подходы к лечению ретинобластомы // Вестник РОНЦ. – 2011. – Т. 22, №2. – С. 41-48.
- Иванов И.А., Левина А.Г. и др. Федеральные клинические рекомендации. Энуклеация, эвисцерация: показания, хирургическая техника, реабилитация // НИИ глазных болезней им.Гельмгольца. – 2015.- 44 с.
- Янченко Т.В, Громакина Т.В, Эпидемиологические аспекты ретинобластомы // Медицина в Кузбассе. - 2015. - Т. 14. – С. 2-4.
Свидетельство о регистрации СМИ ЭЛ № ФС 77-75008 от 1 февраля 2019 года выдано Федеральной службой по надзору в сфере связи, информационных технологий и массовых коммуникаций (Роскомнадзор)
Ретинобластома – эмбриональная детская опухоль сетчатки глаза. Среди всех детских злокачественных новообразований, поражающих глазное яблоко, ретинобластома имеет наибольшее распространение.
На протяжении последнего десятилетия частота выявления данной патологии увеличивается. В настоящее время встречаемость ретинобластомы составляет 1 случай на 10-20 тысяч живых новорожденных.
Шестьдесят процентов выявленных опухолей являются ненаследственными (спорадическими); остальные 40% составляют наследственно-обусловленные формы.
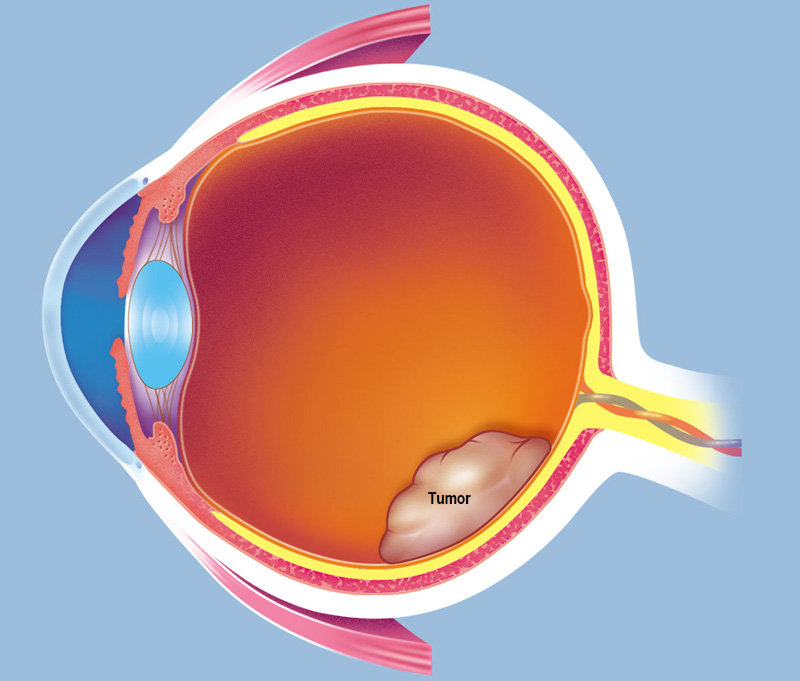
Около 90% случаев ретинобластомы диагностируются до 3 лет. Пик заболевания – 2 года. Заболевание одинаково часто выявляется среди мальчиков и девочек.
Причины. Роль наследственности в заболеваемости ретинобластомой.
- располагается в срединном отделе длинного плеча хромосомы 13q14.1;
- состоит из 27 экзонов;
- занимает 180000 пар нуклеотидов геномной ДНК;
- в норме экспрессируется в клетках всего организма.
Большая часть наследственных и спорадических фактов заболевания обуславливаются делецией в срединном участке длинного плеча хромосомы 13.
В 1971 году Knudson выдвинул гипотезу, согласно которой есть единый механизм формирования ретинобластомы – как наследственной, так и нет, – связанный с инактивацией или утратой двух аллелей гена Rb.
Когда происходит герминальная мутация, все ретинобласты гетерозиготного носителя уже обладают инактивированным аллелем Rb. Чтобы сформировалась ретинобластома, будет достаточно лишь одной телесной мутации, которая затронет оставшуюся копию гена. Переход Rb из гетерозиготного состояния в гомозиготное и является причиной инактивации двух аллелей Rb. Этим и инициируется злокачественное перерождение клетки.
Ненаследственная форма чаще всего проявляется односторонним новообразованием. Ее обнаруживают в 60% случаев. Возникает такая патология в первые 3 года жизни. Данная форма объясняется мутацией в двух аллелях RB1 гена, однако лишь в клетках сетчатки.
Двустороннее поражение может объясняться тем, что в половых клетках родителей присутствует непроявившаяся мутация. Ее они передали ребенку, и возникла болезнь. Также объяснить возникновение такой ретинобластомы можно мутацией de novo, происходящей на ранних стадиях развития эмбриона.
То есть, билатеральные формы патологии при спорадическом типе следует относить к врожденным формам.
Особенности клиники наследственных форм – это:
- ранний возраст заболевающих;
- многофокусность;
- двухстороннее расположение;
- мультицентрический рост новообразования с преобладанием смешанных и экзофитных форм;
- наличие у кого-то в семье такой же опухоли;
- молекулярные и хромосомные аномалии Rb гена.
Если в поколениях не было такого заболевания, характер опухоли односторонний и однофокусный, то можно предполагать, что мутация возникла в родословной впервые.
Симптомы ретинобластомы
По мере того, как новообразование растет и распространяется, формируются: вторичная (на фоне основного заболевания) глаукома, буфтальм (у маленьких детей – увеличение глазного яблока), воспалительные изменения (иридоциклит, увеит). Когда возникает отек орбитальной клетчатки, есть вероятность выпячивания глаза (экзофтальма).
Классификация ретинобластомы
АВС (Амстердам)
В Амстердаме в 2001-м году предложили международную классификацию интраокулярной (внутриглазной) ретинобластомы. Главная цель этой классификации – определить четкие показания к тому, чтобы проводить органосохраняющую терапию. Если поражение двухстороннее, то классификация применяется отдельно к каждому глазу.
Клинические группы, в соответствии с классификацией:
- А – интраретинальные новообразования до 3 мм в размере, располагающиеся в 1,5 мм от диска зрительного нерва и более 3 мм от центральной ямки.
- В – остальные опухоли, располагающиеся отдельно, ограниченные сетчаткой с вероятным наличие субретинальной жидкости в не более чем 3 мм от основы опухоли, не сопровождающееся субретинальным обсеменением (рис. 2).
- С – раздельные опухоли с местным обсеменением стекловидного тела над новообразованием и субретинальным обсеменением в не более чем 3 мм от образования с вероятностью наличия жидкости в субретинальном пространстве в 3-6 мм от основания опухоли.
- D – рассеянные опухоли с крупными опухолевыми массами, разнокалиберными отсевами в стекловидном теле и/или субретинальном пространстве в более чем 3 мм от новообразования с субретинальной жидкостью более 6 мм от образования, включая полную отслойку сетчатки (рис. 3 и 4).
- Е – определяется одним из перечисленных ниже факторов неблагоприятного прогноза:
- новообразование прилегает к хрусталику;
- неоваскулярная глаукома;
- высыхание, утрата функции глаза (фтизис);
- кровоизлияние, ввиду которого утрачена прозрачность стекловидного тела;
- опухоль, располагающаяся кпереди от передней поверхности стекловидного тела и также захватывающая цилиарное тело и переднюю часть глаза;
- некротизированная опухоль с асептическим целлюлитом орбиты;
- ретинобластома в диффузной инфильтративной форме.
![]()
Диагностика ретинобластомы
Применяются следующие клинические методы:
- сбор анамнеза;
- оценка жалоб ребенка;
- физикальный осмотр;
- инструментальные обследования.
Особое внимание врач обращает на наличие наследственного фактора.
Проводится окулистом с медикаментозным расширением зрачка. Врач осматривает глаз с боковым освещением, делает биомикроскопию, офтальмоскопию, тонометрию. Если обследуется совсем маленький ребенок, то применяется наркотический сон.
Является обязательным исследованием, в соответствии со стандартом обследования при таком заболевании. Одной из самых информативных методик признана УЗ-биолокация. Благодаря ей, выявляются плюс-ткани, и это позволяет отличить ретинобластому от отслойки сетчатки, эндофтальмита, фиброза стекловидного тела, ретролентальной фиброплазии.
Чтобы определить величину затухания УЗ в опухоли используют квантитативную эхографию. Для получения информации о динамике злокачественного процесса важное значение имеет УЗ-эхобиометрия. С ее помощью определяют степень проминенции (т. е. то, насколько оно выступающее) новообразования.
МРТ головного мозга и орбит
Магнитно-резонансная томография дает возможность в деталях увидеть распространение процесса за пределы глазного яблока и точно определить стадию болезни. Такое исследование нужно проводить всем пациентам, у которых ретинобластома диагностирована впервые. Контраст при МРТ повышает информативность.
![Визуализация ретинобластомы с помощью МРТ. Клиническая группа В]()
Рис. 2. Визуализация ретинобластомы с помощью МРТ. Клиническая группа В
![Визуализация ретинобластомы с помощью МРТ. Размеры образования соответствуют клинической группе D]()
Рис. 3. Визуализация ретинобластомы с помощью МРТ. Размеры образования соответствуют клинической группе D
![Визуализация ретинобластомы с помощью МРТ. Фронтальное изображение орбит. Клиническая группа D]()
Рис. 4. Визуализация ретинобластомы с помощью МРТ. Фронтальное изображение орбит. Клиническая группа D
Исследование спинномозговой жидкости, костного мозга
Проводится больным, у которых процесс достаточно сильно распространен, чтобы уточнить, есть ли дистантные (удаленные от очага) метастазы.
Цитология цереброспинальной жидкости при прорастании новообразований в субарахноидальное пространство позволяет обнаружить клетки опухоли.
Методы лечения
Сегодня самое пристальное внимание уделяется органосохраняющим методикам. Кроме местной и системной химиотерапии, лучевой терапии, если есть показания, применяются:- фотокоагуляция (удаление новообразования путем воздействия на него мощным световым потоком);
- криодеструкция (разрушение опухоли холодом);
- лазерная деструкция (удаление опухоли лазером).
Принципы органосохраняющей терапии при лечении ретинобластомы
- Больным из группы А проводят только местное лечение (лазерную коагуляцию, криодеструкцию, диатермокоагуляцию).
- Больным из группы В показано 6 курсов двухкомпонентной ПХТ (полихимиотерапии), а также дополнительно – одна из методик местного лечения (лазерная деструкция, криодеструкция, брахитерапия).
- Тем, кто относится к группам С и D, назначаются 6 курсов ПХТ, состоящей из трех компонентов. Лечение дополняют местной химиотерапией и, если существует необходимость, физическими способами деструкции опухоли.
Пациентам из группы Е показана энуклеация (то есть удаление глазного яблока, предполагающее также пересечение наружных глазных мышц и зрительного нерва). В течение 5-7 дней после хирургического вмешательства выполняют глазное протезирование. Подбор постоянного протеза осуществляется в течение 1,5 месяцев после операции.
Энуклеация глаза имеет четкие показания:
- пациент должен относиться к группе Е по амстердамской классификации АВС;
- двусторонняя форма заболевания, необратимая утрата функции одного глаза;
- отсутствие возможности после консервативного лечения оценить степень поражения глаза ввиду катаракты или кровоизлияния в стекловидное тело;
- если поражение обоих глаз зашло слишком далеко и нет шанса восстановить зрение, проводится двусторонняя энуклеация (удаляются оба глаза).
Хирургическое лечение сочетается с ПХТ, облучением, лазерной деструкцией и др.
ПХТ (консервативная терапия) после энуклеации
Показана в следующих случаях:
- прорастание опухоли в зрительный нерв и экстрабульбарное распространение;
- большие или множественные узлы новообразования, локализация опухоли в перипапиллярной зоне, интраокулярная диссеминация (распространение) процесса в радужку, стекловидное тело и др., вовлечение в злокачественный процесс хориоидеи и зрительного нерва.
Если имеет место ретроламинарное распространение образования, то ПХТ дополняется лучевым лечением.
Если опухоль прорастает до линии резекции зрительного нерва и/или имеет место экстрасклеральное распространение, то таких пациентов относят к группе высокого риска. Им показана дистанционная лучевая терапия в сочетании с ПХТ, а затем – высокодозная ПХТ с последующей аутотрансплантацией (пересадкой собственных) периферических стволовых клеток крови.Предлагаем ознакомиться с несколькими наиболее часто применяемыми схемами ПХТ, которые показаны при данном заболевании:
Локальная (местная) химиотерапия
- дозировка 5 мг/м2 показана, когда одновременно лечатся оба глаза;
- если поражен только один, то нужно вводить 7,5 мг/м2.
Физические методы лечения
К ним относятся:
- лазерная деструкция;
- фотокоагуляция;
- криодеструкция;
- транссклеральная диатермокоагуляция.
Рассмотрим данные методики подробно.
Выполняется на аргоне, может применяться для терапии заболевания в 1-й стадии и при небольших опухолевых узлах. При двусторонних ретинобластомах для терапии оставшегося глаза данную методику применяют в комплексе с другими. Наиболее часто ее сочетают с облучением, химиотерапией. Обычно – на последних этапах лечения.
Преимущество такой технологии – выраженная реакция тканей, находящихся вблизи очага поражения. Это позволяет проводить лазерную деструкцию, когда опухоли располагаются рядом со зрительным нервом.
Сочетается с другими методиками (ПХТ, лучевым лечением и др.) на последних этапах терапии. Рекомендуется применять данный метод при 1-й стадии болезни и небольших опухолях: выстояние узла – до 8 мм, поражение глазного дна – до 25% площади.
Методика противопоказана, когда:
- новообразование расположено близко к диску зрительного нерва и макулярной зоне;
- опухоль большая.
Показано применение этой методики в сочетании с фотокоагуляцией. Охлаждающими веществами выступают углекислый газ и жидкий азот.
Преимущество перед диатермокоагуляцией – меньшее повреждение склеры.
Может проводиться в качестве профилактического воздействия после операции. Делается каждый день со 2-3-го послеоперационных дней. Разовая экспозиционная доза – 1,8 Гр, суммарная очаговая – 35-50 Гр. Курс терапии составляет 1-2 серии облучения с интервалом 1,5-2 месяца.
![]()
Наблюдение, объем и сроки обследования
При обнаружении болезни на ранних стадиях выживаемость достигает 100%.
После окончания лечения за детьми с этим заболеванием обеспечивается диспансерное наблюдение у окулиста в поликлинике и в онкодиспансере по месту жительства. Специалист (врач-окулист) должен осматривать ребенка 1 раз в 2 месяца в 1-й год после окончания терапии, а затем:
- во 2-й год – 1 раз в 3 месяца;
- в следующие 2 года – 1 раз в 6 месяцев;
- далее – 1 раз в год.
С такой же периодичностью должны проводиться и осмотры онкологом.
Офтальмологическое исследование проводится с медикаментозным расширением зрачка. Детей младшего возраста обследуют в состоянии наркотического сна.
Если в семье были случаи ретинобластомы, то впервые офтальмолог должен осмотреть ребенка еще в роддоме.
Больные постоянно состоят на учете.
Также под диспансерным наблюдением должны находиться маленькие дети, появившиеся на свет в семьях, в которых есть больные ретинобластомой.
![Ретинобластома: клиника, диагностика и лечение]()
![]()
Авторская публикация:
Иванова Светлана Вячеславовна
Врач-детский онколог, научный сотрудник, кандидат наук
НМИЦ онкологии им Н.Н. Петрова![]()
Под научной редакцией:
Кулева Светлана Александровна
Заведующий отделением, врач- детский онколог, ведущий научный сотрудник, профессор
НМИЦ онкологии им. Н.Н. ПетроваКлючевые слова: дети, ретинобластома, наследственность, ген ретинобластомы, патоморфология, лейкокория, офтальмоскопия, ультразвуковое исследование.
Для цитирования: Иванова С.В., Кулева С.А., Садовникова Н.Н. и др. Ретинобластома. Часть 1. Клинико-диагностические аспекты. РМЖ. Клиническая офтальмология. 2020;20(3):159-164. DOI: 10.32364/2311-7729-2020-20-3-159-164.
Retinoblastoma. Part 1. Clinical presentations and diagnostic tools
S.V. Ivanova 1 , S.A. Kuleva 1 , N.N. Sadovnikova 2 , M.I. Komissarov 2 , M.N. Chistyakova 2 ,
A.V. Khokhlova 1 , N.A. Shchegoleva 31 N.N. Petrov National Medical Research Center of Oncology, St. Petersburg,
Russian Federation2 St. Petersburg State Pediatric Medical University, St. Petersburg, Russian Federation
3 K.A. Rauchfuss St. Petersburg Children’s City Multidisciplinary Clinical Center of High Medical Technologies, St. Petersburg, Russian Federation
Retinoblastoma is one of the most common retinal tumors in young children. At the time of primary diagnosis, mean age is no more than 1.5 years. The association between tumor development and the loss of both alleles of RB1 gene is well-established. Mutations in the RB1 gene result in the complete inactivation of retinoblastoma protein expression and, therefore, uncontrolled cell proliferation and generation of tumor cell clone. There are two types of retinoblastoma, i.e., hereditary and sporadic. This paper describes in detail clinical epidemiological characteristics of hereditary and sporadic retinoblastoma and major clinical signs (in particular, one of the most early and common symptoms, leukocoria). Histology of malignant tumor and its benign precursor is addressed. Current classification systems of intraocular retinoblastoma are based on tumor extent being required to specify clear indications for organ preservation treatment. Modern diagnostic tools for retinoblastoma are discussed as well. Eye fundus examination is the gold standard but has some specifics in young children. Other important imaging techniques in retinoblastoma are sonography, optical coherence tomography, and magnetic resonance imaging.
Keywords: children, retinoblastoma, heredity, retinoblastoma gene, pathomorphology, leukocoria, ophthalmoscopy, sonography.
For citation: Ivanova S.V., Kuleva S.A., Sadovnikova N.N. et al. Retinoblastoma. Part 1. Clinical presentations and diagnostic tools. Russian Journal of Clinical Ophthalmology. 2020;20(3):159–164. DOI: 10.32364/2311-7729-2020-20-3-159-164.
![Ретинобластома. Часть 1. Клинико-диагностические аспекты]()
Введение
Ретинобластома относится к числу наиболее распространенных неоплазий, поражающих орган зрения у детей раннего возраста. Тем не менее заболевание является относительно редким, встречающимся в среднем у одного из 15–18 тыс. новорожденных. Мальчики и девочки поражаются одинаково часто. 70% выявленных опухолей являются спорадическими; остальные 30% представлены наследственно обусловленной формой. Около 95% случаев ретинобластомы диагностируются до 5 лет. В Северной Америке были проанализированы 1452 случая ретинобластомы за период с 1973 по 2009 г. [1]. Средний возраст на момент первичной диагностики не превышал 1,5 года. У пациентов с двухсторонней ретинобластомой заболевание манифестировало раньше и диагностировалось, как правило, на первом году жизни (медиана 0,46 года). Это одно из крупнейших эпидемиологических исследований, представленных в зарубежной литературе. Точных сведений о заболеваемости ретинобластомой в России не существует, поскольку статистическая отчетность формируется на основании кодов МКБ-10, основанных на анатомической локализации поражения, что ведет к искусственному завышению показателей заболеваемости за счет включения других опухолей, поражающих орбиту (лимфомы, саркомы и проч.) [2].
Роль наследственности в заболеваемости ретинобластомой
Конституциональные мутации de novo наиболее часто возникают в половой клетке, обычно мужской. Крайне редко возникает ситуация, когда конституциональная мутация в гене RВ1 происходит через некоторое время после зачатия в одной из нескольких клеток развивающегося эмбриона. В этом случае появляется мозаицизм, и если поражены половые клетки эмбриона, заболевание будет передаваться в поколении [9].
Мутации в гене RВ1 приводят к полной инактивации экспрессии белка ретинобластомы (pRb). Белок-супрессор опухолевого роста pRb является основным ингибитором клеточной пролиферации, регулирующим переход клеточного цикла в S-фазу путем блокировки точки контроля G1. Блокирование перехода G1/S осуществляется, главным образом, через взаимодействие с транскрипционным фактором 1 семейства E2F. В случае мутации белка pRb транскрипционный фактор E2F1 переходит в активное состояние, что в конечном итоге ведет к неконтролируемой пролиферации клетки и формированию клона опухолевых клеток.
Для наследственных форм ретинобластомы характерны ранняя манифестация, билатеральность и мультифокальность поражения, положительный семейный анамнез, а также обнаружение хромосомных и молекулярных аномалий гена ретинобластомы [10]. Заболевание может развиваться как одновременно в обоих глазах, так и метахронно. Наличие герминальной мутации в гене RВ1 способствует повышенному риску развития вторых опухолей: остеогенной саркомы, рака мочевого пузыря, легких, кожи и опухолей мозга на протяжении всей жизни излеченного от ретинобластомы пациента. Фактором, резко повышающим риск развития второй опухоли, является использование дистанционной лучевой терапии. Крайне редко (не более чем в 5% случаев всех наследственных форм) у больных развивается трилатеральная ретинобластома — сочетание двухсторонней ретинобластомы с морфологически идентичной опухолью мозга, наиболее часто локализующейся в шишковидной железе [11].
Спорадическая форма ретинобластомы диагностируется в 70–80% случаев и характеризуется манифестацией в старшем возрасте, унилатеральным и монофокальным характером поражения.
У незначительной доли пациентов (около 3%) развитие ретинобластомы не связано с мутацией в гене RВ1. В последние десятилетия появились работы, свидетельствующие о причастности к возникновению опухоли онкогена MYCN [12, 13]. Заболевание в таком случае характеризуется односторонним поражением и ранней манифестацией в первом полугодии жизни ребенка.
Патоморфология
По гистологической структуре ретинобластома представляет собой злокачественную нейроэктодермальную опухоль, развивающуюся из нервных клеток эмбриональной сетчатки. Опухоль состоит из недифференцированных нейробластических клеток — ретинобластов, которые характеризуются гиперхромными ядрами, скудной цитоплазмой, большим числом митозов. Строма в опухоли отсутствует. В зависимости от степени дифференциации опухолевых клеток различают ретинобластому (встречающуюся чаще) и ретиноцитому.
Согласно Международной классификации заболеваний в онкологии (International Classification of Diseases for Oncology) ретинобластоме присвоен морфологический код 9510/3, а ретиноцитоме — 9510/0 [14].
Ретиноцитома — весьма редкое, одно- или двухстороннее доброкачественное новообразование сетчатки. Ткань опухоли состоит из дифференцированных клеток, из которых образуются истинные розетки Флекснера — Винтерштейнера (рис. 1). Классическая розетка Флекснера — Винтерштейнера представляет собой округлый кластер клеток, сгруппированных вокруг центрального просвета, содержащего мелкие цитоплазматические отростки окружающих клеток. Существует возможность злокачественного перерождения ретиноцитомы, в связи с чем пациенты с этим заболеванием подлежат тщательному динамическому наблюдению.
![Рис. 1. Розетки Флекснера — Винтерштейнера (гематок- силин-эозин, ×400) Fig. 1. Flexner — Wintersteiner rosette (hematoxylin and eosin staining, ×400)]()
Ретинобластома состоит из мелких низкодифференцированных клеток. Различают два типа роста опухоли — эндофитный, или стелящийся, и экзофитный. При эндофитном росте происходит проминирование образования в стекловидное тело, нередко с формированием витреальных отсевов. При экзофитном росте чаще возникает отслойка сетчатки с накоплением субретинальной жидкости, появлением субретинальных отсевов. При распространении опухоли в сосудистую оболочку глазного яблока резко возрастает вероятность гематогенного метастазирования.
В растущей опухоли развиваются новообразованные сосуды. Опухолевые клетки пролиферируют вокруг них, формируя так называемые периваскулярные муфты (рис. 2A). В связи с недостаточным кровоснабжением опухоль рано подвергается некрозу, очаги которого являются источником рассеивания отдельных клеток и целых конгломератов. В очагах некроза откладываются соли кальция, образуя характерные для опухоли кальцификаты (рис. 2В).
![Рис. 2. Микропрепарат ретинобластомы: А — периваскулярные муфты; В – кальцификаты в опухоли (гематоксилин- эозин, ×100) Fig. 2. Microslide of retinoblastoma: A — perivascular invasion; B — calcifications (hematoxylin and eosin staining, ×100)]()
Клиническая картина
По мере роста и распространения опухоли глаз реагирует воспалительным процессом (увеит, иридоциклит). При отеке орбитальной клетчатки и/или распространении в орбиту развивается экзофтальм. При прорастании трабекулярного аппарата и нарушении оттока из глаза внутриглазной жидкости развивается вторичная глаукома, клиническим проявлением которой может стать появление болевого синдрома и (у маленьких детей) буфтальма.
Системы классификации ретинобластомы
По мере расширения представлений о диагностике и лечении ретинобластомы исторически происходила эволюция подходов к классификации этого заболевания.
В 1963 г. А.B. Reese и R.M. Ellsworth предложили классификацию интраокулярной ретинобластомы [15]. Она использовалась вплоть до 2005 г. и имела большое практическое значение в период, когда для лечения широко применялась дистанционная лучевая терапия.
С появлением новых методов лечения в 2005 г. была разработана Международная классификация интраокулярной ретинобластомы (International Intraocular Retinoblastoma Classification — IIRC), которая позволила оценивать прогноз заболевания при применении системной полихимиотерапии и определять четкие показания для проведения органосохраняющего лечения [16]. Согласно классификации пораженные глаза распределяются на клинические группы, которым присваивается буквенная аббревиатура от A до Е (табл. 1). Позднее C.L. Shields et al. (2006 г.) предложили модификацию IIRC, согласно которой в случае поражения опухолью более половины глазного яблока пациент стратифицируется в клиническую группу Е [17]. Таким образом, на настоящий момент в мире одновременно используются как минимум два варианта IIRC, что затрудняет интерпретацию результатов лечения интраокулярной ретинобластомы у разных авторов и не позволяет провести корректное сравнение различных клинических исследований.
![Таблица 1. Международная классификация интраокуляр- ной ретинобластомы IIRC [16] Table 1. International Intraocular Retinoblastoma Classification (IIRC) [16]]()
В 2006 г. группой экспертов, принимавших участие в разработке IIRC, также была предложена унифицированная международная система стадирования экстраокулярной ретинобластомы (International Retinoblastoma Staging System — IRSS), основанная на оценке радикальности выполненной энуклеации и диссеминации опухолевого процесса [18].
Диагностика ретинобластомы
![Рис. 4. Двухсторонняя мультифокальная ретинобластома, группы В (слева) и С (справа) Fig. 4. Bilateral multifocal retinoblastoma, group B (left) and C (right)]()
Данный метод визуализации позволяет дифференцировать ретинобластому от других поражений сетчатки (болезнь Коатса, ретролентальная фибродисплазия, эндофтальмит, гамартома сетчатки и проч.), картировать опухолевые узлы и отсевы, определять степень тяжести поражения глаза и, соответственно, клиническую группу, мониторировать ответ на лечение. Факторами, снижающими диагностическую ценность офтальмоскопии, являются кровоизлияние в стекловидное тело, массивная отслойка сетчатки и непрозрачность сред, нередко обусловленная крупными облаковидными интравитреальными отсевами.
Ультразвуковое исследование глаз также входит в стандарты обязательного офтальмологического обследования при ретинобластоме. Одним из наиболее информативных методов является В-сканирование глазного яблока, позволяющее благодаря визуализации плюс-ткани в различном положении глаза отличить ретинобластому от гемофтальма, эндофтальмита, отслойки сетчатки, ретролентальной фиброплазии, фиброза стекловидного тела и другой патологии органа зрения [20]. Выявление классических кальцификатов в опухоли, без сомнения, подтверждает диагноз ретинобластомы (рис. 5).
![Рис. 6. Оптическая когерентная томограмма глаза с ретинобластомой при эндофитном (А) и экзофитном (В) росте опу- холи (красными стрелками указана сетчатка, белыми стрелками – узел опухоли) Fig. 6. Optical coherence tomography of endophytic (A) and exophyt]()
![Рис. 6. Оптическая когерентная томограмма глаза с ретинобластомой при эндофитном (А) и экзофитном (В) росте опу- холи (красными стрелками указана сетчатка, белыми стрелками – узел опухоли) Fig. 6. Optical coherence tomography of endophytic (A) and exophyt]()
Оптическая когерентная томография позволяет четко визуализировать взаимоотношение опухоли с сетчаткой и хориоидеей, дифференцировать экзо- и эндофитный рост ретинобластомы, дать метрическую характеристику очагам опухоли (рис. 6) [20].
Ультразвуковая биомикроскопия позволяет особенно тщательно оценить передние отделы сетчатки глаза, цилиарную область и передний сегмент [21]. При анализе более чем 100 глаз этот метод не показал ни одного ложноположительного или ложноотрицательного результата, что подтверждает его высокую диагностическую значимость при первичной диагностике и последующем динамическом наблюдении за пациентами [22].
Магнитно-резонансная томография (МРТ) орбит и головного мозга доказала свои преимущества перед компьютерной томографией в оценке распространения опухоли за пределы глазного яблока, инвазии зрительного нерва, поражения вещества головного мозга (метастазы или трилатеральная ретинобластома), она позволяет достаточно точно осуществить стадирование процесса [23, 24]. МРТ-исследование с контрастированием является обязательным методом как на этапе первичной диагностики, так и в процессе динамического наблюдения за пациентами с ретинобластомой.
Заключение
Ретинобластому можно с уверенностью отнести к разряду неоплазий, диагностика которых возможна на ранних стадиях. Несмотря на редкость этого заболевания, практически каждый детский офтальмолог в своей практической работе хотя бы однажды встречает такого пациента. При этом именно офтальмолог играет ключевую роль в установлении диагноза и, что крайне важно, определяет клиническую группу по классификации IIRC. Современные стратегии ранней диагностики опухоли направлены на обеспечение возможности применения органосохраняющих методик в ее лечении, что позволяет сохранить не только жизнь, но и глазное яблоко и зрение у ребенка.
Таким образом, именно содружественная работа детских офтальмологов, детских онкологов и лучевых диагностов является залогом успеха в излечении маленьких пациентов.
Сведения об авторах:
1 Иванова Светлана Вячеславовна — к.м.н., детский онколог отделения химиотерапии и комбинированного лечения злокачественных опухолей у детей, ORCID iD 0000-0002-0585-0907;
1 Кулева Светлана Александровна — д.м.н., заведующая отделением химиотерапии и комбинированного лечения злокачественных опухолей у детей, ORCID iD 0000-0003-0390-8498;
2 Садовникова Наталья Николаевна — к.м.н., заведующая офтальмологическим отделением, ORCID iD 0000-0002-5943-1046;
2 Чистякова Маргарита Николаевна — врач-офтальмолог офтальмологического отделения, ORCID iD 0000-0001-7410-3650;
1 Хохлова Анна Валерьевна — врач-патологоанатом патологоанатомического отделения с прозектурой, ORCID iD 0000-0002-0551-804X;
3 Щеголева Наталья Адольфовна — главный внештатный детский хирург, заместитель главного врача по хирургии, ORCID iD 0000-0003-3672-7319.
2 ФГБОУ ВО СПбГПМУ Минздрава России. 194100, Россия, г. Санкт-Петербург, ул. Литовская, д. 2.
About the authors:
1 Svetlana V. Ivanova — MD, PhD, pediatric oncologist of the Department of Chemotherapy and Combined Treatment of Malignant Tumors in Children, ORCID iD 0000-0002-0585-0907;
1 Svetlana A. Kuleva — MD, PhD, Head of the Department of Chemotherapy and Combined Treatment of Malignant Tumors in Children, ORCID iD 0000-0003-0390-8498;
2 Natal’ya N. Sadovnikova — MD, PhD, Head of Ophthalmological Department, ORCID iD 0000-0002-5943-1046;
2 Margarita N. Chstyakova — MD, ophthalmologist of Ophthalmological Department, ORCID iD 0000-0001-7410-3650;
1 Anna V. Khokhlova — MD, pathologist of Pathoanatomic Department with Prosection, ORCID iD 0000-0002-0551-804X;
3 Natal’ya A. Shchegoleva — MD, Chief Visiting Pediatric Surgeon, Deputy Head Doctor for Surgery, ORCID iD 0000-0003-3672-7319.
1 N.N. Petrov National Medical Research Center of Oncology. 68, Leningradskaya str., Pesochnyy Village, St. Petersburg, 197758, Russian Federation.
2 St. Petersburg State Pediatric Medical University. 2, Litovskaya str., St. Petersburg, 194100, Russian Federation.
3 K.A. Rauchfuss St. Petersburg Children’s City Multidisciplinary Clinical Center of High Medical Technologies. 8A, Ligovskiy av., St. Petersburg, 191036, Russian Fede-
ration.Читайте также:









![Таблица 1. Международная классификация интраокуляр- ной ретинобластомы IIRC [16] Table 1. International Intraocular Retinoblastoma Classification (IIRC) [16]](https://www.rmj.ru/upload/medialibrary/e3f/159-4.jpg)

