
Реферат на тему синдром марфана
Обновлено: 02.07.2024
Кононова Анастасия
Преподаватель: Кириченко Н.В.
2. История изучения . 5
3. Клинические проявления . …. 6
4.Лечение и профилактика. 8
Список литературы . 11
ВВЕДЕНИЕ
Среди всех наследственных заболеваний соединительной ткани наибольший интерес для терапевтов и врачей общей практики представляет синдром Марфана, так как продолжительность жизни этих больных без лечения ограничена 30–40 годами и у одного пациента может быть огромное количество проблем со здоровьем. Поскольку заболевание имеет заведомо серьезный прогноз для жизни и трудоспособности пациентов, установление диагноза накладывает особую ответственность на врача при первой встрече с больным.
1. ОПИСАНИЕ
Синдром (болезнь) Марфана - аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани. Установлено, что причиной патологии является мутация гена фибриллина FBN 1. Заболевание полиморфно – может протекать с разной выраженностью клинической картины, и характеризуется появлением все новых типов мутаций в генах. Синдром Марфана получил своё название от фамилии французского педиатра А. Марфана, который вперые представил описание 5-летней девочки Габриель с необычными, непрерывно прогрессирующими аномалиями скелета, и дал патологии своё имя. Распространенность синдрома — 1 случай на 10000 человек. Риск рождения ребенка с синдромом Марфана повышается после достижения отцом возраста 35 лет и достигает 50% при наличии патологии у одного из родителей. Врожденная аномалия наследуется по аутосомно-доминантному типу. В ее основе лежит дефект важнейшего гена, отвечающего за синтез коллагена. Во время внутриутробного развития происходит нарушение формирования волокон соединительной ткани, утеря ими прочности, в результате чего волокна не способны выдерживать естественные нагрузки. Поэтому наибольшие атипичные изменения претерпевают крупные сосуды, клапаны сердца, связки глаза, твердое небо, скелет и мышцы.
Без адекватной терапии продолжительность жизни людей с синдромом Марфана не более 40 лет. Терапия позволяет увеличить этот срок вдвое и более.
2. ИСТОРИЯ ИЗУЧЕНИЯ
В 1876 г. симптомы неизвестной патологии были отмечены доктором Вильямсом, но клинические наблюдения проводились гораздо позже — в 1896 г. педиатром из Франции А. Марфаном. Врач в течение 5-ти лет оценивал состояние девочки с неизученными ранее аномалиями, заключающимися в прогрессировании дистрофии скелета и мышечной ткани. К середине 20-го века имелось множество описанных случаев, когда у больных наблюдались симптомы, близкие к патологии Марфана, и все они относились к заболеваниям наследственного типа. Среди таких случаев — расслоение аорты, пороки сердца, эктопия хрусталиков, сопровождающиеся деформацией костей (грудной клетки, позвоночника) и внешними отклонениями от нормы (высокий рост, худоба, длинные конечности). Американским генетиком МакКьюсиком было проведено детальное исследование мутаций хромосом и открыта новая группа заболеваний соединительной ткани.
3. КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ
Частый признак синдрома Марфана — высокий рост (до 200 см.), при этом туловище непропорционально короткое, а конечности удлиненные и тонкие. Пальцы у больных длинные, паукообразные (арахнодактилия). Из-за недоразвития подкожной клетчатки и мышечной дистрофии страдающие синдромом Марфана имеют астеническое телосложение.
Другие внешние симптомы патологии (в каждом индивидуальном случае может наблюдаться один или несколько из них):
— вывихи шейного сегмента позвоночника;
— деформация грудной клетки;
— глубокая посадка глаз;
— уменьшенная нижняя челюсть, нарушение роста зубов;
— паховые грыжи, частые разрывы связок.
Более серьезные изменения при синдроме Марфана протекают в организме. Самые тяжелые из них развиваются со стороны сердца и сосудов и могут привести к смерти ребенка еще на первом году жизни. Среди них:
— дефекты ветвей легочной артерии, аорты (расширения, аневризмы, расслоения);
— пороки сердца (чаще — поражения клапанов);
Подобные нарушения вызывают тахикардию, мерцательную аритмию вплоть до фибрилляции предсердий или развития сердечной недостаточности.
Со стороны глаз наблюдается выраженная миопия, вывих хрусталика, аномалии развития роговицы, уменьшение в размерах радужки, косоглазие, патологии сосудистой стенки сетчатки. При прогрессирующем вывихе хрусталика или при отслойке сетчатки уже в раннем возрасте больные могут полностью потерять зрение.
Со стороны нервной системы при синдроме Марфана происходит растяжение твердой мозговой оболочки и выбухание ликвора в костные дефекты в пояснично-крестцовом отделе позвоночника (дуральная эктазия).
Легкие страдают гораздо реже, так как незначительные нарушения их работы не оказывают влияния на дыхательную функцию. Но в отдельных случаях снижение эластичности альвеол может привести к спонтанному пневмотораксу, развитию дыхательной недостаточности.
Кожные покровы. Отмечаются атрофичные стрии, не связанные с колебаниями веса, беременностью или физическими растяжениями, рецидивирующие грыжи любой локализации.
Прочими симптомами патологии могут быть эктопия почек, деформации мочевого пузыря, половых органов.
4. ЛЕЧЕНИЕ И ПРОФИЛАКТИКА
Специфической терапии заболевания не существует: изменить гены еще до рождения ребенка невозможно. Лечение только симптоматическое и зависит от тех изменений в организме, которые развиваются у больного синдромом Марфана . Некоторые осложнения патологии можно успешно корректировать, другие — устранять оперативным путем.
Пациент должен наблюдаться у группы специалистов — офтальмолога, невролога, кардиолога, ортопеда, хирурга. Основное направление терапии — поддержка функций сердца и сосудов.
Методы лечения :
— прием препаратов (адреноблокаторы, антиаритмические лекарства, антикоагулянты и т.д.);
— хирургия пороков сердца (дисфункции клапанов, расширения, расслоения легочной артерии), аорты, протезирование клапанного аппарата.
Нормализация зрения проводится при помощи коррекции миопии (ношение очков, линз), лечения катаракты, глаукомы, имплантации искусственного хрусталика.
При поражении суставов и позвоночника проводится оперативное лечение (протезирование, пластика суставов, устранение межпозвоночных грыж), выправление кифоза, сколиоза при помощи тракции, мануальной терапии. Из медикаментозных средств используются миорелаксанты, витамины группы В. Также применяется физиолечение, занятия ЛФК.
При поражении легких часто требуется хирургическое вмешательство (дренирование их полости).
Беременность больными синдромом Марфана должна строго планироваться и развиваться под контролем группы врачей, специализирующихся на лечении людей с подобными патологиями. Родоразрешение — только при помощи кесарева сечения. Еще до наступления беременности желательно обследоваться на предмет возможного прогрессирования расслойки аорты и, по возможности, провести операцию по замене части сосуда. Консультация генетика позволит рассчитать примерный риск по передаче заболевания по наследству.
При наблюдении за больными с синдромом Марфана необходимо выполнять следующие требования к режиму труда, отдыха и реабилитации:
1. Детям с синдромом Марфана разрешаются занятия физкультурой только по ослабленной программе (спецгруппы и группы ЛФК);
2. Категорически запрещаются занятия в спортивных секциях, участие в соревнованиях, сельскохозяйственных работах, походах на длительные дистанции по пересеченной и горной местности, ношение тяжестей (не более 3 кг);
3. Категорически запрещены специальности, связанные с профессиональной вредностью: контакты с химическими веществами, лаками, красками, работа в условиях высоких температур и воздействия радиации, а также профессии, сопряженные с вибрацией, требующие высокой остроты зрения, больших физических и эмоциональных затрат;
4. При выборе места жительства больным противопоказаны жаркий климат и зоны повышенной радиации;
5. Беременным женщинам с синдромом Марфана необходимо один раз в 2 месяц проводить эхокардиографию. При диаметре аорты 45 мм и выше следует безотлагательно решать вопрос о целесообразности дальнейшего сохранения беременности;
6. Родоразрешение женщин с синдромом Марфана необходимо осуществлять с помощью кесарева сечения в специализированных родильных домах для рожениц с патологией сердечно-сосудистой системы.
ПРИЛОЖЕНИЕ
Внешний вид ребенка, больного синдромом Марфана
СПИСОК ЛИТЕРАТУРЫ
1. Засухина Т.Д., Львова Т.Н., Васильева И.Т. Пониженная способность к репарации повреждений ДНК, индуцированных мутагенами в клетках больных синдромом Марфана. // Доклады АН СССР, 1982. — 265. — 5. — сс. 1261 — 1263.
2. Лисиченко О.Ф. Синдром Марфана. / Новосибирск, Наука, 1986. — 163 с.
3. Прозоровская Н.Н., Глинянная С.В., Геращенко Л.П. и др. Влияние терапии бета-адреноблокатором и комплексом витаминов на показатели экскреции оксипролина при некоторых наследственных болезнях соединительной ткани // Вопросы медицинской химии, 1988. — т. 35. — № 5. — с. 99 — 104
5. Н.П.Бочков. Клиническая генетика:учебник,3-е изд., испр. И доп.-М.: ГЭОТАР –МЕД,2004. с.170-173.
6. Н.П.Шабалов. Детские болезни: учебник,5-е изд.,т.2-СПб: Питер,2002. с.482-484.
7. Семячкина А.Н., Сельверова Н.Б., Любченко Л.Н. Гормональные нарушения при болезни Марфана. В сборнике: Наследственные нарушения роста и развития у детей. Москва 1993, с.55–63.
8. Яковлев В.М., Дубилей Г.С. Восстановительное лечение при дисплазии соединительной ткани. Омск: Изд–во ОГМА 1996. 120с.
Синдром Марфана (англ. Marfan syndrome, болезнь Марфана) - аутосомно-доминантное генетическое заболевание которое поражает соединительную ткань, характеризующееся диспропорционально длинными конечностями, тонкими худыми пальцами, соответственно худым телосложением и наличием сердечно-сосудистых пороков, которые специфически проявляются в виде пороков сердечных клапанов и аорты. Это генетическое заболевание связано с нарушением функционирования соединительной ткани и значительным полиморфизмом клинических проявлений.
Это генетическое заболевание связано с нарушением функционирования соединительной ткани и значительным полиморфизмом клинических проявлений.
Вложенные файлы: 1 файл
реферат Cиндром Марфана.doc
Cиндром Марфана (Болезнь Марфана)
Синдром Марфана (англ. Marfan syndrome, болезнь Марфана) - аутосомно-доминантное генетическое заболевание которое поражает соединительную ткань, характеризующееся диспропорционально длинными конечностями, тонкими худыми пальцами, соответственно худым телосложением и наличием сердечно-сосудистых пороков, которые специфически проявляются в виде пороков сердечных клапанов и аорты. Это генетическое заболевание связано с нарушением функционирования соединительной ткани и значительным полиморфизмом клинических проявлений.
Это генетическое заболевание связано с нарушением функционирования соединительной ткани и значительным полиморфизмом клинических проявлений.
Преимущественно эта болезнь наследуется по доминантному признаку и вызывается аномалией гена FBN1, кодирующего белок фибрилин-1. У каждой личности есть пара таких генов. Поскольку наследование происходит по доминантному типу, то люди, что наследуют один аномальный ген FBN1 от кого либо из родителей будут поражены указанным заболеванием. Синдром Марфана может появляться в умеренной и тяжелой формах. Люди с этим заболеванием, как правило, высокие, с длинными конечностями и длинными худыми пальцами. Наиболее серьезными осложнениями болезни является повреждение клапанов сердца и нарушение структуры стенок аорты. Также заболевание может влиять на легкие, глаза, твердую оболочку спинного мозга, скелет и твердое нёбо.
Кроме функций связующего белка, который служит опорой для ткани за пределами клетки, белок фибрилин связывается с другим белком, вследствие чего образуется трансформирующий фактор роста бета (TGF-β). TGF-β имеет негативное влияние на сосудистый тонус гладких мышц и нарушает развитие целостного внеклеточного матрикса. Еще одной причиной развития болезни вследствие мутации гена ответственного за синтез фибрилина, сегодня ученые называют накопление избыточного количества TGF-β в легких, клапанах сердца и в аорте, что ослабляет ткани и вызывает симптомы болезни Марфана. Поскольку блокаторы рецепторов ангиотензина уменьшают количество TGF-β, что было продемонстрировано с помощью фармакологических средств, блокирующих функцию этих рецепторов (напр. лозартан и др.). В небольшом клиническом исследовании с участием молодых людей с тяжелой формой болезни Марфана, применявшие блокаторы рецепторов к ангиотензину II у некоторых пациентов действительности рост аорты существенно сократился, и соответственно риск тяжелых сердечнососудистых осложнений снизился.
Болезнь получила название от имени Антуана Марфана, французского педиатра, который впервые описал симптомы заболевания в 1896 году, заметив черты синдрома у пятилетней девочки. Ген, который вызывает болезни был впервые обнаружен Франческо Рамиресом в центре Маунт Синай (Нью-Йорк) в 1991 г.
Хотя нет никаких уникальных симптомов болезни Марфана, однако, сочетание таких признаков как длинные конечности, дислокация хрусталика, аневризма корня аорты, вполне достаточно для того, чтобы с уверенностью поставить диагноз. Однако, более чем 30 других клинических признаков, связанные с исследуемым синдромом. Большинство из них характеризуются изменениями скелета, кожи и суставов. Существует также сравнительно высокая вероятность того, что в разных семьях наблюдаться идентичные мутации.
Поражение костной системы
Большинство видимых признаков болезни Марфана, связанные с костной системой. У многих людей с синдромом Марфана рост значительно выше среднего. Некоторые из них имеют длинные конечности с длинными тонкими пальцами рук и ног (арахнодактилия). Кроме диспропорций развития конечностей и чрезмерного роста болезнь Марфана вызывает и другие нарушения в функционировании костной системы. Особенно распространены искривление позвоночника (сколиоз), воронкообразная (внутрь) и килевидная (наружу) деформация грудной клетки, чрезмерная гибкость суставов, высокое небо, неправильный прикус, плоскостопие, молоткообразная деформация пальцев стопы (когда суставы пальцев на ногах согнуты и напоминают, поэтому молоток, сутулость, появление беспричинных растяжек на коже (стрии). У некоторых пациентов может появляться боль в суставах, костях и мышцах. Также у больных с синдромом Марфана иногда возникают расстройства или нарушения речи (через высокое небо и малые размеры челюсти). Ещё существует вероятность развития остеоартрита в раннем возрасте.
Нарушение функций глаз и зрения
Болезнь Марфана может влиять на зрение и глаза. Обычно у пациентов наблюдаются астигматизм и близорукость, однако иногда фиксируется и дальнозоркость. Нарушение положения хрусталика в одном или обоих глазах (эктопия хрусталика), наблюдается у 80% больных. Выявить эти проблемы со здоровьем может офтальмолог (окулист) с помощью щелевой лампы (метод биомикроскопии). При синдроме Марфана дислокация преимущественно имеет суперотемпоральний характер (вверх и наружу), тогда как при подобном состоянии гомоцистеинемии - инфероназальний (вниз и внутрь). Иногда проблемы со зрением возникают только после ослабления соединительной ткани, которое вызвано расслоением сетчатки. Еще одной из офтальмологических проблем, связанных с синдромом Марфана можно назвать раннюю (в молодом возрасте) глаукому.
Наиболее серьезными признаками и симптомами болезни Марфана, является нарушение деятельности сердечно-сосудистой системы. Чрезмерная усталость, отдышка, нарушения ритма сердца, тахикардия (учащенное сердцебиение), стенокардия (которая сопровождается возникновением болевых ощущений в спине, плече или руке) - это те нарушения, которые наблюдаются при синдроме Марфана. Часто у пациентов, из-за нарушения кровообращения конечности (руки, ноги) холодные. Причинами для дальнейших обследований такого пациента может быть присутствие шумов в сердце, изменения на ЭКГ (электрокардиограмма) или присутствие симптомов стенокардии. Одной из причин регургитации (движения крови в противоположную сторону нормального направления), которая возникает из-за пролапса аортального или митрального клапанов сердца, можно назвать кистозную медиальную дегенерацию клапанов, которая возникает из-за влияния болезни Марфана.
Однако основным признаком для дальнейшего детального исследования и изучения заболевания является расширенная аорта или аневризма аорты (выпячивание стенки аорты). Однако, иногда, очевидных проблем с сердечной системой не наблюдается, но ослабление соединительной ткани (через кистозную дегенерацию средней стенки сосудов - медии) вызывает аневризму или расслоения восходящей части аорты, что требует хирургического лечения. Расслоение аорты часто сопровождается болями в спине или груди и приводит к возникновению ощущения надрыва. Через нарушение функциональности соединительной ткани (что является патогенетическим механизмом развития синдрома Марфана) увеличивается частота случаев, при которых искусственный митральный клапан не приживается в организме больного. Именно поэтому необходимо проявлять чрезвычайную осторожность при лечении клапанов сердца. Предпочтение следует отдавать таким мерам, которые направлены на восстановление функциональности больного клапана, а не на немедленную его замену.
Во время беременности у женщин с синдромом Марфана, даже при отсутствии видимых отклонений в работе сердечно- сосудистой системы, высокий риск расслоения аорты, что может привести к летальному исходу, даже при своевременном лечении. Именно поэтому, если у женщины присутствует это заболевание, то перед зачатием необходимо пройти тщательное исследование и получить консультацию врача. А во время самой беременности каждые шесть-десять недель нужно проводить эхокардиографию для определения диаметра корня аорты. Во многих случаях возможны естественные роды без осложнений, однако, лишь после исчерпывающего медицинского обследования и оценки всех возможных рисков.
Влияние на лёгкие
Болезнь Марфана является одним из факторов риска для спонтанного возникновения пневмоторакса, при котором воздух выходит из легких и занимает плевральную полость между грудной клеткой и легкими, в результате чего легкие сжимаются. Больной пневмотораксом испытывает резкую боль в груди, дышит часто и поверхностно, наблюдается выраженная отдышка. Часто проявляется бледность или синюшность кожных покровов, в частности лица (цианоз). Если заболевание не лечить, оно может привести к смерти больного.
Кроме этого, синдром Марфана может быть связан с такими заболеваниями легких как апноэ во сне (это прекращение вентиляции легких во время сна, более чем на 10 секунд) и другими идиопатическими (с неустановленной причиной) обструктивными болезнями легких.
Влияние на центральную нервную систему
Одним из последствий болезни Марфана, который может негативно повлиять на качество жизни человека (хотя он не представляет угрозы жизни) является дуральная ектазия. Это ослабление и растяжение твердой оболочки мозга, а точнее соединительной ткани дурального мешка - мембраны, которая окутывает спинной мозг.
В течение длительного времени симптомы дуальной ектазии (боль в пояснице, в ногах, в области живота и таза, другие неврологические симптомы в нижних конечностях или головная боль) могут не проявляться. Или же резко исчезают, когда человек лежит на плоской ровной поверхности, на спине. При болях такого типа врачи обычно назначают рентген поясничного отдела позвоночника, хотя, как правило, дуральную ектазию невозможно заметить с помощью рентгена на ранних стадиях. Именно поэтому, ухудшение симптомов и отсутствие, какой либо другой причины боли создает необходимость проведения исследования с помощью магнитно резонансной томографии (МРТ) поясничного и крестцового отделов позвоночника. Дуральную ектазию, которая вызывает такие симптомы, будет хорошо видно на вертикальном изображении МРТ. Она будет иметь вид расширенных отростков, которые направлены к поясничным позвонкам. Другие неврологические проблемы, связанные с синдромом Марфана - это дегенеративные заболевания междупозвоночных дисков и костей спины. Также синдром Марфана является важным фактором вызывающим развитие дисфункции автономной нервной системы.
Синдром Марфана вызывается мутациями в гене FBN1 (15 хромосома), который кодирует гликопротеин фибрилин-1, являющийся компонентом внеклеточного матрикса. Белок фибрилин-1 имеет важное значение для правильного формирования внеклеточного матрикса, играет определенную роль при биогенезе и влияет на функционирование эластичных волокон. Кроме того, внеклеточный матрикс обеспечивает структурную целостность соединительной ткани, и играет роль резервуара для факторов роста (класс небольших природных пептидов и белков), главной целью которых является стимулирование роста клеток. Эластиновых волокон очень много во всем организме человека, но сконцентрированы они в основном в аорте, связках, в частности в цинновой связке (особая связка, с помощью которой хрусталик прикрепляется к цилиарному телу), именно поэтому эти части организма повреждаются при болезни Марфана больше всего. Для изучения механизма развития болезни Марфана была взята так называемая трансгенная мышь в организме которой в единственном экземпляре находился мутированный фибрилин-1 (мутации были аналогичны тем, которые происходят при изменении гена, кодирующего этот гликопротеин), который, как известно, является причиной развития синдрома Марфана. Этот вид мышей позволит изучить патогенез синдрома Марфана, ведь их черты болезни аналогичны человеческим. У мышей снижение нормального уровня фибрилина-1 приводит к развитию расстройств, связанных с болезнью Марфана.
Синдром Марфана существенно влияет на трансформирующий фактор роста бета (TGF-β). Ведь фибрилин-1, косвенно связывает неактивную форму TGF-β, будто поглощая ее, что в свою очередь приводит к снижению биологической активности этого фактора. Согласно простейшей схеме развития болезни Марфана, можно предположить, что снижение уровня фибрилина-1 приводит к увеличению количества TGF-β- из-за недостаточного его поглощения. И, хотя не доказано, что повышение уровня TGF-β приводит к возникновению патологий, связанных с синдромом Марфана, но, как известно, вследствие воспалительной реакции освобождения протеаз происходит постепенное ухудшение функциональности эластиновых волокон и других компонентов внеклеточного матрикса.
Достоверность данной гипотезы было подтверждена после открытия подобного синдрома с названием Синдром Лойса-Дитца (Syndrome Loeys-Dietz), который развивается при участии гена TGF-βR2 (3 хромосома), кодирующего рецептор к TGF-β. Через схожие клинические признаки эти два заболевания часто путают между собой.
Критерии, согласно которым диагностируют болезнь Марфана, были согласованы на международном уровне в 1996 году. Диагностика синдрома Марфана базируется на историях семей и на сочетании основных и второстепенных признаков болезни, которые в совокупности редко встречаются среди населения, но в единичном варианте могут проявляться в отдельной личности. Например - четыре нарушения со стороны опорно-двигательного аппарата сочетаются с патологиями других органов (органов системы зрения или сердечно-сосудистой системы) у одного человека - что может быть вероятным признаком синдрома Марфана.
Ниже перечисленные признаки могут быть вызваны болезнью Марфана, или могут возникнуть у людей, в которых любые нарушения отсутствуют:

Синдром Марфана — наследственное заболевание соединительной ткани с аутосомно-доминантным типом наследования. В результате мутации нарушается синтез фибриллина, что ведет к аномальному строению α-цепи коллагена I типа, а также эластина, которые являются структурным компонентом сердечных клапанов, сердечной мышцы, стенок сосудов и опорно-двигательного аппарата [3].
На данный момент известно больше 500 мутаций гена фибриллина-1 (FBN1), но также заболевание может быть связано с деформациями генов FBN2, FBN3. Чаще всего возникают нарушения в процессе связывания кальция с фибриллином, в результате чего утрачивается устойчивость фибриллина к протеазам.
Около 15 лет назад был выделен синдром Марфана 2 типа, который обусловлен мутацией гена рецептора 2 трансформирующего ростового фактора β1. Клинически заболевание проявляется скелетными деформациями и расстройствами сердечно-сосудистой системы [3].
Клинические проявления синдрома Марфана могут быть как ярко выраженными, так и стертыми.
Люди, страдающие данным заболеванием, имеют высокий рост, арахнодактилию, скелетные деформации, патологии органа зрения, кардиоваскулярные патологии [4]. Часто новорожденные дети страдают недостаточностью клапанов сердца, дилатацией проксимальной части аорты. Нарушается формирование эластического каркаса аорты и аортальных клапанов с некрозом и фрагментацией эластических волокон, разрушаются коллагеновые волокна, происходит дистрофия гладкомышечных клеток с накоплением в них мукополисахаридов, разрастаются кисты, что приводит к острой сердечно-сосудистой недостаточности. В этом случае продолжительность жизни ребенка редко превышает один год [2].
Один из ведущих симптомов синдрома Марфана — поражение органа зрения (за исключением синдрома Марфана второго типа, где в клиническом статусе превалируют нарушения со стороны опорно-двигательного аппарата, сердечно-сосудистые расстройства, но отсутствуют поражения глаз) [1,3]. Патологоанатомически это определяется слабостью связочного аппарата хрусталика, что может привести к его вывиху. Также отмечаются слишком малые размеры и неправильная форма хрусталика, вследствие чего могут возникать деструкции и даже разрывы связок, что, в свою очередь, приводит к эктопии [1]. Еще одним симптомом является уменьшение диаметра сосудов, питающих сетчатку. Гетерохромия, мегалокорнеа, катаракта, гипоплазия радужной оболочки, цилиарной мышцы, пигментной каймы зрачков выявляются в 3 случаях из 10. Увеличение длины оси глазного яблока влечет за собой миопию и в дальнейшем может вызывать отслойку сетчатки [3].
На МРТ и КТ головы у более чем половины больных определяется диффузное утолщение твердой мозговой оболочки, дивертикулы паутинной, истончаются рога спинного мозга. При этом стоит отметить, что перечисленные выше заболевания прогрессируют с возрастом, что подтверждает динамическое наблюдение больных с синдромом Марфана [4].
Синдром Марфана классифицируют в зависимости от течения, генетических характеристик и клинических вариантов. Автором одной из классификации является Н. Т. Ватутинов [1].
.
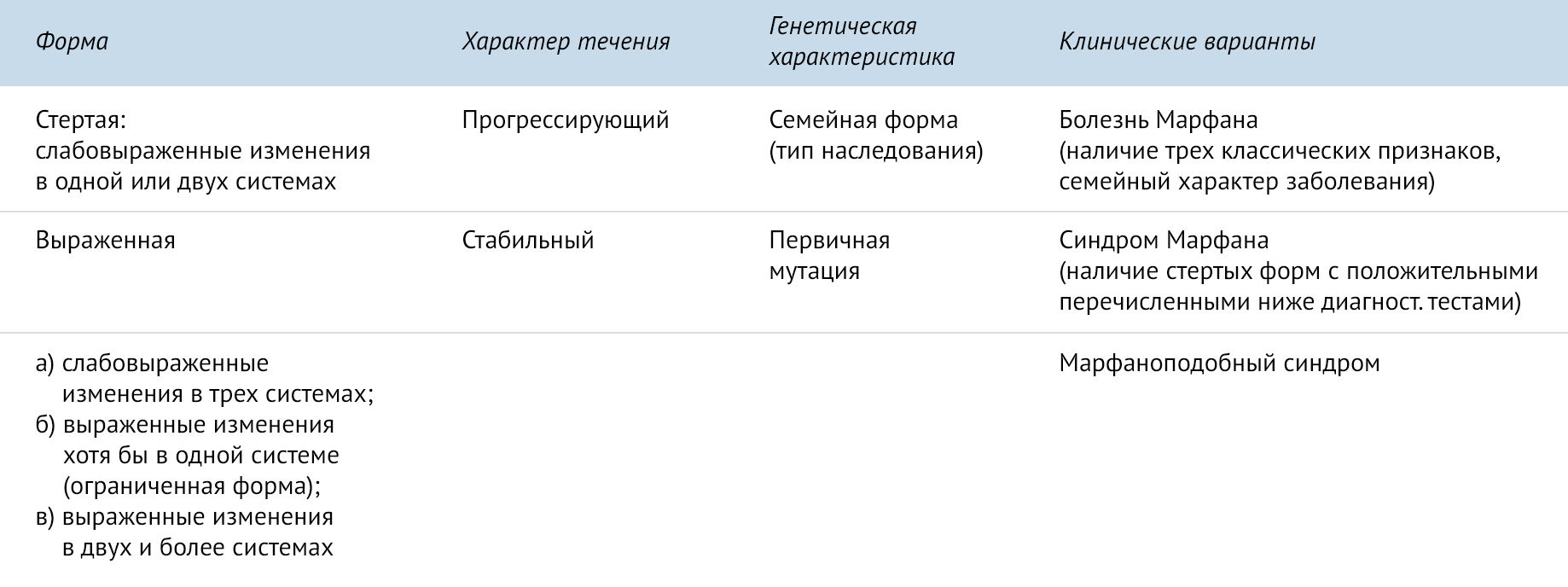
Данные критерии были пересмотрены, и в 1989 году были приняты так называемые Гентские критерии. Для постановки диагноза необходима совокупность двух больших критериев по двум системам и одного малого критерия по третьей системе [1].
.
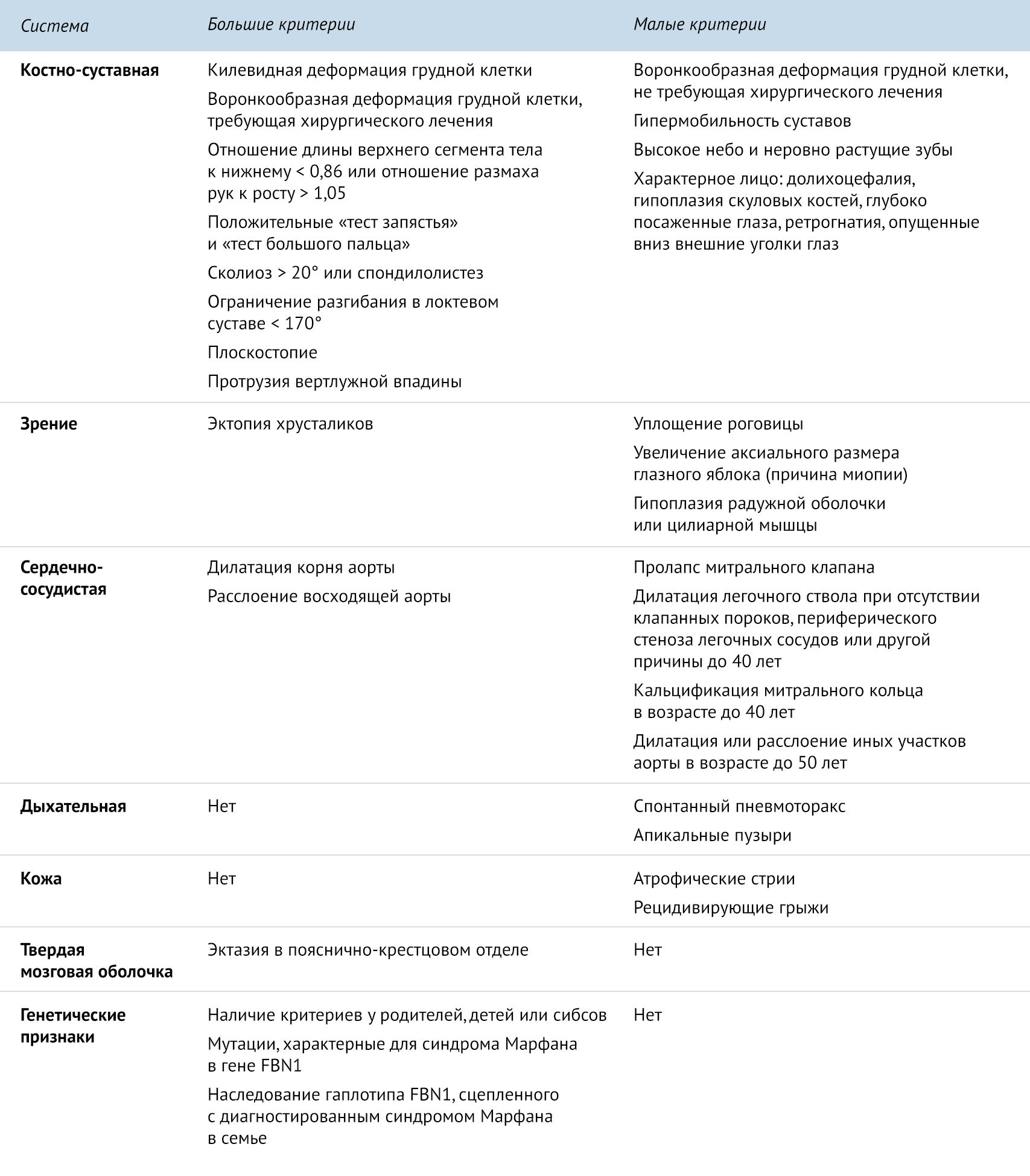
Синдром Марфана необходимо дифференцировать с аутосомно-рецессивным заболеванием — гомоцистинурией, типичным проявлением которого является повышенный уровень метионина в крови и гомоцистина в моче, что не встречается при синдроме Марфана [1].
.
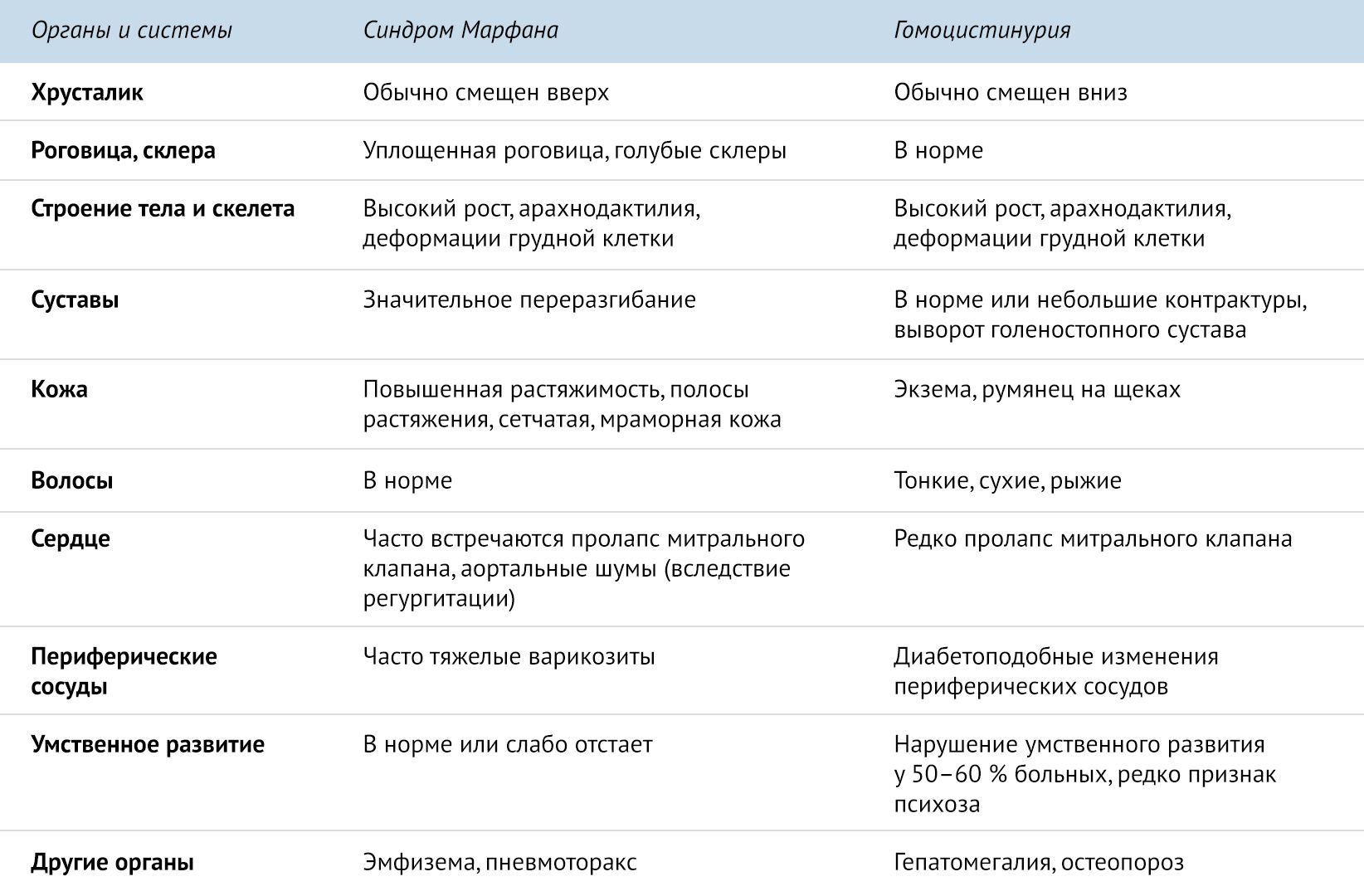
Несмотря на то что синдром Марфана влечет за собой достаточно серьезные нарушения, пациенты, страдающие данным недугом, вполне способны поддерживать нормальное функционирование своих органов и систем в течении достаточно длительного времени. Для этого используются упражнения, направленные на укрепление мышц спины и брюшного пресса, а также связочного аппарата. Важным является и то, что занятия физкультурой позволяют поддерживать нормальный психический статус пациентов, а также стимулируют и укрепляют ССС. Показатели рекомендуемых нагрузок для таких пациентов составляют 25–50 Вт с увеличением ЧСС не более 110 уд/мин.
Лечение преимущественно направлено на общее укрепление организма [4].
Обязательным является сочетанием физических упражнений с общеукрепляющей медикаментозной терапией. Назначаются курсами по 2 месяца такие препараты, как терафлекс, диоксихолекальциферол, оксидевит, Кальций-Д3 Никомед. Обязательная терапия направлена на обогащение организма кальцием, магнием, фосфором, селеном. Лечение проводится с постоянным мониторированием уровня кальция и фосфора в крови и моче. Биохимический контроль показателей крови и мочи рекомендуется проводить не реже 1 раза в 2 недели. С целью улучшения усвоения аминокислот назначают анаболики [1].
Реабилитационная терапия проводится курсами не менее двух раз в год при контроле биохимических маркеров, таких как экскреция оксипролина (предшественник коллагена) и гликозаминогликанов с суточной мочой, уровень оксипролина и лизина в сыворотке крови, а также содержание кальция и фосфора в крови и моче, активность щелочной фосфатазы в крови. Диетотерапия проводится при наличии у больных поверхностного гастродуоденита, патологических рефлюксов, нарушения моторики на фоне повышения кислотообразующей функции желудка. Постоянный рацион питания должен быть богат продуктами, содержащими витамины А, Е, С, макро- и микроэлементы — кальций, медь, магний, железо, цинк, фосфор [1,4].

Актуальность: Синдром Марфана одно из самых распространенных наследственных заболеваний, которое сложно диагностировать, полностью излечиться от него нельзя, можно только снимать симптомы и в некоторых случаях прибегать к хирургическому вмешательству, приступим к изучению данного заболевания.
Синдром (болезнь) Марфана(СМ) — это аутосомно-доминантное заболевание относящееся к группе наследственных патологий соединительной ткани. Проявляется в результате мутации гена, который кодирует синтез гликопротеина фибриллина-1, также является плейотропным. Свойственна различная экспрессивность и пенетрантность.
Имеет большой фенотипический набор, бывают легкие формы, трудно отличимые от нормы, но есть и тяжелые, это объясняется разнообразием мутаций в гене FBN 1, а также наличие мутаций в иных генах. В результате генетических исследований было установлено, что в 75%-ом случае синдром Марфана наследуется семейно, в остальных случаях - первичная мутация. Риск рождения ребенка с синдромом зависит от возраста отца, чем старше - тем больше риск.
Цель работы: изучить этиологию, эпидемиологию, симптомы и лечение Синдрома Марфана.
Эпидемиология.
Синдром Марфана - это редкое заболевание, для которого характерно классическое менделевеское наследование. Распространенность в популяции составляет порядка 1 на 5000. Синдром диагностируется везде, независимо от этнической группы.
У больных фенотип характеризуется определённой протяжённостью, начиная от лёгких форм соединительнотканной дисплазии, которые встречаются и в общей популяции - до случаев с угрозой для жизни с системными расстройствами.
Мышечно-скелетная система: арахнодактилия, долихостеномелия, деформации позвоночника (сколиоз, гиперкифоз и т.д.), деформация передней стенки грудной клетки, плоская стопа, высокое готическое нёбо, гипермобильность суставов, врождённые контрактуры локтей и пальцев, мышечная гипотония, недоразвитие вертлужной впадины.
Сердечно-сосудистая система: пролапс митрального клапана отмечается в 80 % случаев; дилатация корня аорты начинается с синуса Вальсальвы и прогрессирует с возрастом (у женщин отмечается более медленное прогрессирование) и в конечном итоге может приводить к расслаивающейся аневризме аорты.
Иные системы органов: у 5 % больных наблюдаются спонтанные пневмотораксы ; характерны стрии на коже в областях плеч, груди, поясницы; у большей части больных наблюдается сужение нервного канала в пояснично-крестцовом отделе; довольно таки часто диагностируются кистозные образования в печени и почках, которые увеличиваются с возрастом и, как правило, клинически не значимы.
Лечение - в основном симптоматическое, направленное на облегчение проявлений заболевания. Больным требуется ежегодно проходить медицинское обследование с обязательным участием кардиолога, ортопеда и офтальмолога.
Большая часть клинических исследований поддерживает профилактическое применение бета-адреноблокаторов с раннего возраста для предотвращения расслаивающейся аневризмы аорты . Если ярко выражена дилатация корня аорты выполняется его хирургическая правка. Главным симптомом для операции у взрослых является достижение максимального диаметра корня аорты 50 мм.
С целью предотвращения инфекционного эндокардита и тромбозов после хирургических операций назначают антибиотики и коагулянты.
При синдроме Марфана проводят коррекцию зрения с помощью подбора очков и линз, если присутствует необходимость можно применить лазерное или хирургическое лечение катаракты, удаление смещенного хрусталика и имплантация искуственного. При наличии ярко выраженных скелетных нарушений может потребоваться хирургическая стабилизация позвоночника, торакопластика и т. п. Также применяют патогенетическую коллагеннормализующую терапию, метаболическую и витаминотерапию.
В данной работе мною было изучены этиология, эпидемиология, симптомы и лечение Синдрома Марфана. Синдром Марфана серьёзное заболевание его сложно диагностировать. Длительность жизни больных напрямую зависит от того насколько сильно проявились сердечно - сосудистые изменения, а также поражения скелета и глаз. Однако кардиохирургия способствует остановке роста аорты и её расслоения, это позволяет продлевать жизни больным с Синдромом Марфана. Больным необходимо находиться под постоянным контролем специалистов и регулярно проводить диагностические обследования.
Список литературы:
1) Лисиченко О. В. Синдром Марфана. Новосибирск: Наука, 1986. 164 с.
3) Фищенко Я.В. (2006) Алгоритм диагностических проявлений синдрома Марфана.
Читайте также: