
Приобретенная апластическая анемия реферат
Обновлено: 02.07.2024
Приобретённая (идиопатическая) апластическая анемия – панцитопения с гипоцеллюлярным костным мозгом в отсутствие патологической инфильтрации и повышения количества ретикулиновых волокон.
Апластическая анемия – гетерогенное заболевание. В большинстве случаев (70-80%) этиология остается неясной – идиопатическая апластическая анемия. Примерно в 15-20% наблюдаются конституциональные/врожденные варианты апластической анемии. В некоторых случаях удается установить причину апластической анемии – лекарственное воздействие (таблица 1), инфекцию и др.
Таблица 1. Препараты, использование которых может быть ассоциировано с АА.
| Группа | Препараты |
| Антибиотики | Хлорамфеникол, Сульфаниламиды, Ко-тримоксазол, Линезолид |
| Противовоспалительные препараты | Препараты золота, Пеницилламин, Индометацин, Диклофенак, Напроксен, Пироксикам, Сульфасалазин |
| Противосудоржные препараты | Карбамазепин, Фенитоин |
| Тиростатики | Тиоурацил, Карбимазол |
| Антидепрессанты | Фенотиазины |
| Гипогликемические средства | Хлорпропамид, Толбутамид |
| Противомалярийные препараты | Хлорохин |
| Другие | Аллопуринол, Тиазиды |
Код(ы) МКБ-10:
| МКБ-10 | |
| Код | Название |
| D 61.3 | Идиопатическая апластическая анемия |
Дата разработки/пересмотра протокола: 2015 год (пересмотр 2018 г.)
Сокращения, используемые в протоколе:
полимеразная цепная реакция
Пользователи протокола: врачи общей практики, терапевты, гематологи, гинекологи.
Категория пациентов: взрослые, беременные.
Шкала уровня доказательности [1]:

АА может носить как наследственный, так и приобретенный характер. Несколько редких врожденных заболеваний, включая анемию Фанкони, синдром Швахмана-Даймонда, врожденный дискератоз, первично характеризуются апластическим типом гемопоэза [2].
Известно, что некоторые препараты также могут вызывать селективные цитопении и агранулоцитоз, которые обычно обратимы и проходят после прекращения воздействия агента. Эти обратимые реакции не коррелируют с риском апластической анемии, ставя под сомнение эффективность рутинного мониторинга клинического анализа крови в качестве метода диагностики апластической анемии [1].
Исследования in vitro и данные клинических наблюдений привели к выводу, что основой для большинства случаев приобретенной апластической анемии служит атака цитотоксическими Т-лимфоцитами CD34+ клеток и стволовых клеток крови (СКК). Иммунное повреждение клеток костного мозга после лекарственной, вирусной или токсической аплазии костного мозга может быть результатом индукции неоантигенов, провоцирующих вторичную Т-опосредованную атаку на кроветворные клетки. Спонтанное или митоген-индуцированное увеличение продукции мононуклеарами интерферона-γ, IL-2 и фактора некроза опухолей-α (TNF-α) ингибирует дифференцировку гемопоэтических клеток [1]. Секреция интерферона-γ является результатом повышения регуляции транскрипционного фактора T-bet и апоптоза CD34+ клеток, частично опосредованного FAS-зависимым путем [3].
В ранних лабораторных экспериментах удаление лимфоцитов из апластического костного мозга увеличивало число колоний клеток в культуре тканей, а их внедрение в нормальный костный мозг подавляло гематопоэз in vitro. Эффекторные клетки были идентифицированы при помощи иммунофенотипирования активированных цитотоксических CD8+ Т-клеток, экспрессирующих Th1-цитокины, в частности γ-интерферон. CD8 клетки могут определяться непосредственно в крови при помощи:
- проточной цитометрии T-клеточного рецептора (TCR);
- спектрального анализа комплемент-определяющего региона (CDR2);
- секвенирование области CDR3 для установления молекулярного клона.
Также снижение количества регуляторных Т-клеток (CD4+, CD25+, FoxP3+) способствует увеличению аутореактивной популяции CD8+ CD28– Т-клеток, которая индуцирует апоптоз аутологичных гемопоэтических клеток. Т-регуляторные клетки являются компонентами иммунной системы, подавляющими иммунные реакции других клеток, а также играющими роль в предотвращении аутоиммунных реакций [2].
АА может возникать из-за метаболической или иммунологической предрасположенности (полиморфизма генов) у восприимчивых лиц. В случае фенилбутазон-ассоциированной аплазии костного мозга происходит замедление процессов окисления и клиренса соответствующего соединения, ацетанилида, по сравнению с нормальной контрольной группой, что предполагает избыточное накопление лекарственного средства как потенциального пускового механизма развития аплазии.
Для пациентов как с приобретенной, так и с наследственной апластической анемией (Фанкони, врожденный дискератоз) характерно наличие дефекта теломераз и восстановления теломер. Одной из характерных черт лейкоцитов при апластической анемии является укорочение теломер, причиной чему предполагали деплеции стволовых клеток. Однако анализ наследования в больших родословных продемонстрировал, что X-связанная форма врожденного дискератоза была вызвана мутациями в DKC1 (dyskeratosis congenita 1), идентификация мутаций в TERC (Telomerase RNA Component) у пациентов с аутосомно-доминантным наследованием помогла выявить генетическую основу укорочения теломер [3].
Центральную роль в восстановлении структуры РНК играет шаблон РНК, кодируемый TERC, при этом теломераза, представляющая собой обратную транскриптазу, кодируемую TERT (Telomerase Reverse Transcriptase), удлиняет нуклеотидную последовательность; другие белки, включая дискерин, кодируемый DKC1, связаны с восстановлением теломер. Систематические исследования ДНК выявили мутации TERC и TERT у некоторых пациентов с приобретенной апластической анемией. У членов семей, имеющих данную мутацию, несмотря на нормальные или почти нормальные показатели крови, выявили уменьшенное количество CD34-клеток, плохую скорость образования гемопоэтических колоний, повышенный уровень гемопоэтического фактора роста, гипоцеллюлярные участки в костном мозге, и, конечно, короткие теломеры. Клиническое проявление этих аномалий проявлялось позже, чем при типичном дискератозе, и не вызывало характерных физических отклонений. Некоторые из пациентов с синдромом Швахмана-Даймонда (Shwachman-Bodian-Diamond) также имеют мутации гена SBDS.
АА может сосуществовать или, по-видимому, эволюционировать вместе с другими гематологическими заболеваниями, характеризующимися пролиферацией специфических клонов клеток, такими как пароксизмальная ночная гемоглобинурия (ПНГ) или миелодиспластический синдром (МДС). Наличие небольшого количества клонов также создает проблемы при постановке диагноза апластической анемии даже при использовании таких высокочувствительных методов диагностики, как фенотипический (проточная цитометрия для ПНГ) или цитогенетический (флуоресцентная гибридизация in situ (FISH) для MDS) анализ [1, 2].
Конечным результатом иммуноопосредованного повреждения костного мозга является уменьшение образования клеток крови в костном мозге. Количество CD34+ клеток и их производных у пациентов с апластической анемией заметно снижено [2].
Клинически AA не сопровождается увеличением лимфатических узлов, печени или селезенки. Основным проявлением заболевания является панцитопения: для клинического анализа крови характерно уменьшение содержания всех форменных элементов. На ранних стадиях можно наблюдать изолированную цитопению, чаще тромбоцитопению. Может присутствовать моноцитопения, что требует дифференциального диагноза с волосатоклеточным лейкозом. АА сопровождается снижением ретикулоцитарного индекса, относительное число ретикулоцитов обычно меньше 1 % и может быть равно нулю, абсолютное число ретикулоцитов — менее 40 000 в мкл (40×10 9 /л), несмотря на высокие уровни эритропоэтина; анизоцитоз и пойкилоцитоз отсутствуют. Эти изменения в периферической крови сопровождаются гипоклеточностью костного мозга без аномальных или злокачественных клеток или фиброза. Необходимо тщательное исследование мазков крови для исключения наличия диспластических клеток. При АА может быть увеличено содержание фетального Hb: у детей это требует проведения дифференциального диагноза с миелопролиферативными миелодиспластическими синдромами, такими как ювенильный миеломоноцитарный лейкоз или другие подтипы МДС [3, 4].
Критерии диагностики АА:
- концентрация гемоглобина (Hb) 9 /л;
- количество нейтрофилов 9 /л;
- содержание ретикулоцитов 9 /л;
- фрагменты костного мозга в аспирате — гипоцеллюлярные с жировым замещением;
- отсутствие диспластических мегакариоцитов и бластных форм; их присутствие указывает на гипопластическую МДС либо эволюцию лейкемии
АА подразделяется на:
- Нетяжелую: отсутствие признаков тяжелой АА [4].
- Тяжелую: клеточность костного мозга 9 /л;
— число тромбоцитов 9 /л;
— количество ретикулоцитов 9 /л.
- Очень тяжелую: те же признаки, что и при тяжелой, но количество нейтрофилов 9 /л;
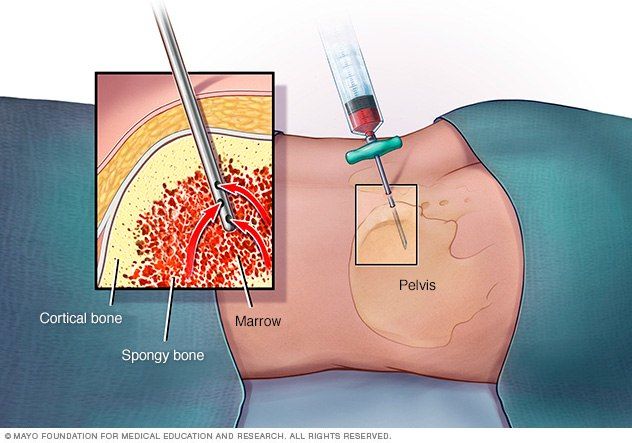
Основным методом диагностики является проведение трепанобиопсии. Аспират костного мозга при АА обычно содержит трабекулы с пустыми, заполненными жиром межтрабекулярными пространствами и малым количеством гемопоэтических клеток. Могут присутствовать единичные лимфоциты, плазматические клетки, макрофаги и тучные клетки [1].
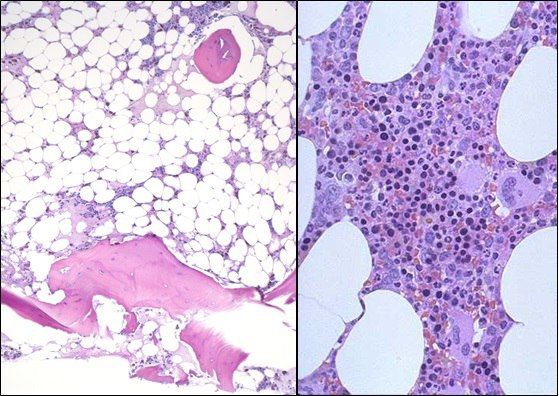
Слева — апластическая анемия, справа — норма
При выполнении цитогенетического анализа могут возникнуть трудности из-за низкой клеточности материала: для получения достаточного количества клеток может потребоваться несколько аспираций. Обнаружение клональных цитогенетических аномалий при апластической анемии является признаком наличия миелоидного заболевания. Переход к более новым методам, таким как сравнительная геномная гибридизация (CGH), позволяет обнаруживать анеуплоидии, делеции, дублирование и/или амплификации любого локуса.
Терапия проводится при помощи следующих препаратов и методов:
Также необходима поддерживающая терапия при помощи гемотрансфузии. Единого целевого значения концентрации гемоглобина не существует, и оно должно быть установлен индивидуально. Для снижения риска иммунизации следует учитывать фенотип Rh и Kell. Пациентам с АА, получающим активное лечение и находящимся в стабильном состоянии, следует назначать профилактические переливания тромбоцитов, пороговая концентрация тромбоцитов составляет 10×10 9 /л. У пациентов, имеющих факторы риска кровотечения, число тромбоцитов должно составлять 20×10 9 /л. Регулярные профилактические переливания тромбоцитов не рекомендуются для стабильных пациентов с АА, не находящихся на активном лечении в данный момент. У пациентов с апластической анемией при регулярной поддержке переливания РБК будет развиваться перегрузка тканевого железа. Сывороточный ферритин остается наиболее широко используемым показателем для оценки перегрузки железом. Магнитно-резонансная томография может определять содержание железа количественно и является хорошим дополнением к лабораторным методам мониторинга [4].
Апластическая анемия – угнетение функции кроветворения красного костного мозга (эритроцитопоэза, лейкопоэза и тромбоцитопоэза), приводящее к пангемоцитопении. К основным клиническим проявлениям гематологического синдрома принадлежат головокружение, слабость, обмороки, одышка, покалывание в груди, кожные геморрагии, кровотечения, склонность к развитию инфекционно-воспалительных и гнойных процессов. Заболевание диагностируется на основании характерных изменений гемограммы, миелограммы и гистологического исследования трепанобиоптата. Лечение патологии включает проведение гемотрансфузий, иммуносупрессивной терапии, миелотрансплантации.
МКБ-10



Общие сведения
Апластическая (гипопластическая) анемия – тяжелое расстройство гемопоэза (чаще всех его звеньев), сопровождающееся развитием анемического, геморрагического синдромов и инфекционных осложнений. Развивается в среднем у 2 человек на 1 млн. населения в год. Приблизительно с одинаковой частотой патология поражает мужчин и женщин. Возрастные пики заболеваемости приходятся на возраст 10–25 и старше 50 лет. При данной патологии в костном мозге чаще нарушается образование всех трех типов клеточных элементов крови (эритроцитов, лейкоцитов и тромбоцитов), иногда - только одних эритроцитов; в зависимости от этого различают истинную и парциальную апластическую анемию. В гематологии данный вид анемии относится к числу потенциально фатальных заболеваний, приводящих к гибели 2/3 заболевших.
Причины
По происхождению апластическая анемия может быть врожденной (связанной с хромосомными аберрациями) и приобретенной (развившейся в течение жизни). Принято считать, что угнетение миелопоэза связано с появлением в красном костном мозге и крови цитотоксических T-лимфоцитов, производящих фактор некроза опухолей и γ-интерферон, которые в свою очередь подавляют ростки кроветворения. Запускать этот механизм могут различные внешнесредовые (химические соединения, физические явления, лекарственные вещества), а также эндогенные факторы (вирусы, аутоиммунные реакции). К числу наиболее значимых причин относят:
- Прием миелотоксических препаратов. Достоверно установлена связь анемии с приемом некоторых противоопухолевых, противосудорожных, антибактериальных, антитиреоидных, противомалярийных препаратов, транквилизаторов, препаратов золота и др., обладающих потенциальным миелотоксическим эффектом. Лекарственные вещества могут вызывать как прямое повреждение стволовых кроветворных клеток, так и опосредованное - через аутоиммунные реакции. Анемии, связанные с таким механизмом развития, называются лекарственными.
- Контакт с химическими и физическими агентами. Супрессию костного мозга может вызывать взаимодействие с органическими растворителями, соединениями мышьяка, бензольными соединениями, пестицидами, облучение всего тела. В некоторых случаях недостаточность гемопоэза является временной и обратимой - главными факторами здесь являются концентрация/доза вещества и время контакта. супрессию костного мозга.
- Вирусные инфекции. Из вирусных агентов наибольшее значение уделяется возбудителям гепатитов В, С и D. В этом случае гипопластическая анемия обычно развивается в течение полугода после перенесенного вирусного гепатита. При изучении патогенеза было замечено, что репликация вируса происходит в мононуклеарах крови и костного мозга, а также в иммунных клетках. Предполагается, что подавление миелопоэза в этом случае является своеобразным иммунным ответом, возникающим против клеток, несущих на своей поверхности вирусные антигены. Такой вид анемии выделяется в отдельную форму – постгепатитную. Среди других вирусных инфекций называются ЦМВ, инфекционный мононуклеоз, грипп.
Также описаны случаи панцитопении, вызванные инфицированием туберкулезом, интоксикацией, лучевой болезнью, лимфопролиферативными заболеваниями (тимомой, лимфомой, хроническим лимфобластным лейкозом), беременностью. Почти в половине наблюдений причину анемии выявить не удается - такие случаи относят к идиопатической форме.
Патогенез
В основе апластической анемии может лежать либо первичное повреждение гемопоэтических стволовых клеток, либо нарушение их эффективной дифференцировки. При наследственных анемиях недостаточность гемопоэза опосредована кариотипическими аберрациями, приводящими к нарушению репарации ДНК и невозможности репликации стволовых клеток костного мозга. В случае приобретенной анемии под влиянием этиофакторов наблюдается активация Т-клеток, которые начинают продуцировать цитокины (интерферон-гамма, ФНО), поражающие клетки-предшественники гемопоэза. В стволовых клетках костного мозга повышается экспрессия генов, отвечающих за апоптоз и активизацию клеточной гибели. Основные клинические проявления обусловлены пангемоцитопенией – снижением в составе крови всех ее форменных элементов (эритроцитов, лейкоцитов, тромбоцитов).
Классификация
Кроме различных этиологических вариантов (лекарственного, постгепатитного, идиопатического), различают острую (до 1 мес. течения), подострую (от 1 до 6 мес.) и хроническую (более 6 мес.) форму заболевания. Анемию, протекающую с избирательным угнетением эритропоэза, называют парциальной красноклеточной аплазией. На основании выраженности тромбо- и гранулоцитопении данная форма анемии подразделяется на 3 степени тяжести:
- очень тяжелую (тромбоцитов менее 20,0х109/л; гранулоцитов менее 0,2х109/л)
- тяжелую (тромбоцитов менее 20,0х109/л; гранулоцитов менее 0,5х109/л), по данным трепанобиопсии – низкая клеточность костного мозга (менее 30% от нормы)
- умеренную (тромбоцитов более 20,0х109/л; гранулоцитов более 0,5х109/л)
Симптомы апластической анемии
Поражение трех гемопоэтических ростков (эритро-, тромбоцито- и лейкопоэза) обусловливает развитие анемического и геморрагического синдромов, инфекционных осложнений. Дебют апластической анемии обычно происходит остро. Анемический синдром сопровождается общей слабостью и утомляемостью, бледностью кожи и видимых слизистых, шумом в ушах, головокружением, покалыванием в груди, одышкой при нагрузке.
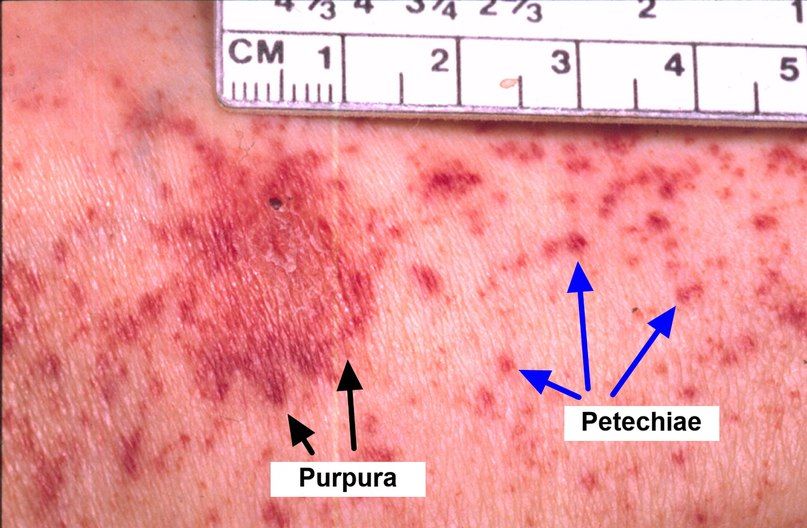
Основным проявлением тромбоцитопении выступает геморрагический синдром. Больные отмечают появление петехий и экхимозов на коже, повышенную кровоточивость десен, спонтанные носовые кровотечения, меноррагии. Возможно возникновение гематурии, маточных и желудочно-кишечных кровотечений. Следствием лейкопении и агранулоцитоза служит частое развитие инфекционных процессов – стоматитов, пневмоний, инфекций кожи и мочевыводящих путей. Для апластической анемий нехарактерны похудание, лимфаденопатия, гепато- и спленомегалия – при этих признаках следует искать другую причину пангемоцитопении.
Врожденная апластическая анемия (синдром Фанкони) обычно развивается у детей в возрасте до 10 лет и кроме аплазии костного мозга характеризуется другими нарушениями: микроцефалией, гипоплазией почек, низкорослостью, аномалиями развития верхних конечностей (гипоплазией первой пястной и лучевой кости), гипоспадией, гиперпигментацией кожи, крайней степенью тугоухости и др. При наследственной анемии Эстрена-Дамешека отмечается тотальное поражение кроветворения и панцитопения при отсутствии врожденных аномалий развития. Для анемии Даймонда-Блекфена или парциальной красноклеточной аплазии характерно только снижение количества эритроцитов.
Осложнения
Летальный исход может быть обусловлен кровоизлияниями во внутренние органы, массивными кровотечениями, инфекционными осложнениями, анемической комой. Наиболее грозное из геморрагических осложнений – кровоизлияние в головной мозг (геморрагический инсульт). Больные склонны к частым и тяжело протекающим вирусным и бактериальным инфекциям респираторного тракта. Значительное или стремительное снижение уровня красных кровяных телец может привести к анемической коме. При молниеносной форме крайне быстро развиваются тяжелейшая анемия, иммунодефицит, коагулопатии, имеющие фатальные последствия.
Диагностика
Оценка гематологического статуса включает внимательный клинический осмотр и проведение тщательной лабораторной диагностики. При физикальном обследовании выявляется выраженная бледность или желтушность кожи, артериальная гипотония, тахикардия. Основу диагностического алгоритма составляет проведение общего и биохимического анализа крови, стернальной пункции, трепанобиопсии:
- Исследования крови. Для гемограммы при гипопластической анемии типичны эритро-, лейкоцито- и тромбоцитопения, нейтропения и относительный лимфоцитоз. Оценка биохимических показателей (печеночных проб, нефрологического комплекса, сывороточного железа, билирубина) информативна для исключения других анемий.
- Исследованиепунктата костного мозга. В миелограмме обнаруживается уменьшение количества миелокариоцитов и мегакариоцитов, снижение клеточности. В трепанобиоптате определяется замещение красного костного мозга жировым (желтым).
В рамках диагностического поиска апластическую анемию необходимо дифференцировать с мегабластными (В12-дефицитными, фолиеводефицитными) анемиями, идиопатической тромбоцитопенической пурпурой, пароксизмальной ночной гемоглобинурией, острым лейкозом.
Лечение апластической анемии
Больные с апластической анемией госпитализируются в специализированные отделения. Им обеспечиваются полная изоляция и асептические условия для предупреждения возможных инфекционных осложнений. Проведение эффективного лечения является сложной проблемой практической гематологии. В зависимости от уровня цитопении используются следующие лечебные подходы:
- Иммуносупрессиная терапия. При умеренной цитопении назначается фармакотерапия, включающая комбинацию антитимоцитарного иммуноглобулина и циклоспорина А. Поддерживающая терапия проводится анаболическими стероидами или их сочетанием с циклоспоринами.
- Гемотрансфузии. В комплексе с курсом иммуносупрессивной терапии при низких показателях красной крови показано проведение заместительной гемотрансфузионной терапии (переливание тромбоцитов и эритроцитарной массы), плазмафереза. Данная мера не оказывает воздействия на патогенетическое звено заболевания, но позволяет восполнить дефицит кровяных телец, не вырабатываемых костным мозгом.
- Трансплантация КМ и СК. Наиболее благоприятные прогнозы на долгосрочную выживаемость оказывает выполнение аллогенной трансплантации костного мозга. Однако ввиду сложности подбора иммунологически совместимого донора процедура используется ограниченно. В качестве экспериментальных подходов рассматриваются аутологичные трансплантации, пересадка стволовых клеток периферической крови. Больным с нетяжелой формой анемии может быть показано проведение спленэктомии, эндоваскулярной окклюзии селезеночной артерии.
Прогноз и профилактика
Прогноз определяется этиологической формой, тяжестью и остротой течения анемии. Критериями неблагоприятного исхода служат быстрое прогрессирование заболевания, тяжелый геморрагический синдром и инфекционные осложнения. После трансплантации костного мозга ремиссии удается достичь у 75–90% пациентов. Первичная профилактика данной разновидности анемии предполагает исключение влияния неблагоприятных внешнесредовых факторов, необоснованного применения лекарственных препаратов, предупреждение инфекционной заболеваемости и др. Пациентам с уже развившимся заболеванием требуется диспансерное наблюдение гематолога, систематическое обследование и длительная поддерживающая терапия.
2. Комплексная программа диагностики апластической анемии с определением прогностически значимых патогенетических особенностей заболевания. Методические рекомендации. - 2015.
4. Апластическая анемия: современные представления о патогенезе и терапии/ Айсариева Б. К., Раймжанов А. Р., Айтбаев К.// Молодой ученый. - 2011 - №9.

Апластической анемией (AA) называют заболевание кровяной системы, которое характеризуется глубокой панцитопенией, которая развивается из-за угнетения костномозговоro кроветворения . В числе главных механизмов поражения кроветворения при апластической анемии является иммунная агрессия, обращенная на клетки-предшественники гемопоэза.
Эпидемиология
AA - уникальная болезнь, которая, с частотностью всего несколько раз на один миллион, бывает у взрослого населения. АА обитает практически у всех возрастных категориях , но, несмотря на это, особенно выделяются две группы - в десять - двадцать пять лет и в преклонном возрасте, обычно шестидесяти лет. Еще одна особенность - встречается у мужчин реже, чем у женщин.
Этиология и патогенез
У пятидесяти процентов зараженных причины появления болезни неизвестны. Обычно это называется идиопатической формой болезни.
Известны несколько причин риска, которые индуцируют появление и развитие апластической анемии:
химические соединения : дозозависимые ( в определенной дозе наносят повреждения в костном мозге, нарушая образование клеток крови ) -цитостатики, иммунодепрессанты;
вещества, действие которых на костный мозг изъясняется индивидуально высокой чувствительностью больного - антибиотики, нестероидные противовоспалительные препараты, сульфаниламиды, ртуть, препараты золота, бензин, бензол, нитроэмали, пестициды;
К главным патогенетическим факторам апластической анемии относят:
поражение полипотентной стволовой гемо - поэтической клетки;
поражение клеточного микроокружения стволовой
гемопоэтической клетки и опосредованное нарушение ее функции;
иммунная депрессия и индукция апоптоза стволовых кроветворных клеток;
укорочение жизни эритроцитов;
нарушение метаболизма кроветворных клеток.
Точкой поражения кроветворения при апластической анемии является имунная агрессия, которая направлена на клетки - предшественники гемоэпоза. Также при АА может развиться костномозговая недостаточность. Она возникает из-за подавления пролиферации гемопоэтических клеток-предшественников активированными Т-лимфоцитами и естественными киллерами.
К нарушению процессовов пролиферации и к стимуляции аптоза приводят активация Т-лимфоцитов, выброс медиаторов имунной супрессии кроветворения, экспансия цитотоксических Т-клонов . Из-за этого сильно уменьшается количество пулагемопоэтических клеток и развитие аплазии костного мозга.
Классификация
Обычно ученые по этиологии разделяют наследственные и приобретенные формы АА.
1.Наследственная гипопластическая анемия с поражением трех кроветворных ростков :
с наличием врождённых аномалий развития (анемия Фанкони); без врождённых аномалий развития (анемия Эстрена-Дамешека);
2.Наследственная парциальная анемия с избирательным поражением эритропоэза (анемия Джозефса-Блекфена-Даймонда);
3. Врожденный дискератоз.
Приобретенная анемия с поражением всех трёх кроветворных ростков;
Приобретённая парциальная анемия с избирательным поражением эритропоэза :
Идиопатическая ( форма Эрлиха);
Также разделяют по такому критерию, как степень тяжести:
тяжёлую апластическую анемию - определяется при наличии двух любых из ниже перечисленных критериев периферической крови: гранулоциты 9 /л, тромбоциты 9 / л, ретикулоциты 9 /л;
нетяжелую АА - гранулоцитопения > 0,5х10 9 /л.
Клиническая картина
Обычно почти все симптомы апластической анемии можно интегрировать в 3 главных синдрома: геморрагический, анемический и синдром инфекционных осложнений. Резкое начало апластической анемии встречается у пятнадцати процентов зараженных и характерно появлением носовых, десневых и маточных кровотечений.
Геморрагическому синдрому характерно появление кровоизлияний на коже и слизистых оболочках, носовых
кровотечений, кровотечений из десен, почечных кровотечений и кровоизлияний в головной мозг.
У зараженных агранулоцитозом могут развиваться язвенный стоматит,пневмонии, гингивит, синусит.
Гемолитическому синдрому у больных апластической анемией характерны несколько иные признаки: желтуха, увеличение содержания непрямого билирубина, уменьшение продолжительности жизни эритроцитов; при внутрисосудистом гемолизе возможно появление гемосидеринурии,
гемоглобинурии, повышение содержания в крови свободного гемоглобина, появление в моче уробилина, отсутствие билирубина.
Диагностика
В общем анализе крови определяется выраженное снижение гемоглобина и количества эритроцитов; анемия нормохромная,
нормоцитарная характерно отсутствие или резкое снижение ретикулоцитов, наблюдается лейкопения за счёт гранулоцитопении с относительным лимфоцитозом, характерна тромбоцитопения. Скорость оседания эритроцитов обычно увеличена.Содержание сывороточного железа повышено, процент насыщения трансферрина существенно увеличен.
В миелограмме наблюдается выраженное уменьшение клеток эритроцитарного и гранулоцитарного рядов, редукция мегакариоцитарного ростка, количество ретикулоцитов снижено, появляется сидероцитоз и сидеробластоз.
Гистологическое исследование трепанобиоптата гребня подвздошной кости обнаруживает сильное снижение количества костномозговых кроветворных элементов и замещение кроветворного мозга жировой тканью.
Диагноз апластической анемии ставится только лишь на основании результатов лабораторного исследования и клинических проявлений: Трехростковая цитопения: анемия (гемоглобин 9 /л), тромбоцитопения(тромбоциты 9 /л);
снижение клеточности костного мозга и отсутствие мегакариоцитов по данным пунктата костного мозга;
аплазия костного мозга в биоптате подвздошной кости, преобладание жирового костного мозга.
Дифференциальная диагностика
Апластическую анемию необходимо отличать от болезней, которые обычно сопровождаются цитопенией: миелодиспластическим синдромом (МДС), острым лейкозом, болезнь Маркиафавы-Микеле(при развитии АА с гемолозом) ,агранулоцитозом.
Для МДС гипоплазией также присущи панцитопения и опустошение костного мозга, но в масках крови встречаются и незрелые нейтрофилы или ядросодержащие эритроциты. В кроветворных клетках костного мозга можно обнаружить диспластические изменения, а цитогенетическое исследование обнаруживает клональные хромосомные аномалии.
При Маркиафавы-Микеле характерны гемосидеринурия и гемоглобинурия, высокий уровень свободного гемоглобина в плазме.
Общие принципы лечения
Однако эффективность вылечивания апластической анемии повышается при появлении совершенствованной иммуносупрессивной терапии. Но длительная выживаемость зараженных тяжёлой апластической анемией все так же невысока: всего лишь семьдесят - восемьдесят процентов переживают пять лет.
Эффективность лечения напрямую связана с тяжестью заболевания. Также она тесно связана и с возможностью реализации программы комбинированной иммуносупрессивной терапии или трансплантации костного мозга (ТКМ) при первых стадиях болезни. ТКМ показана в следующих случаях: наличие гистосовместимого донора костного мозга, молодой возраст пациента, короткий гемотрансфузионный анамнез и тяжёлая форма болезни.
Однако огромным минусом этого метода считается ограниченная возможность применения, которое обычно связано с отсутствием донора костного мозга у большого количества больных. Однако, ТКМ теперь считается терапией выбора на первом этапе вылечивания юных пациентов тяжёлой апластической анемией только, если он имеет гистосовместимого родственного донора костного мозга.
Главным средством лечения апластической анемии считается программная иммуносупрессивная терапия. Это система лечебных процедур, которые проводятся этапами в течение всего года и даже больше. Она включает в себя антитимоцитарный глобулин ( АТГ), циклоспорин А (ЦсА), по необходимости (рефрактерной АА) - повторные курсы АТГ и прочие процедуры иммуносупрессивной терапии, позволяющие добиться дольшей выживаемости больных.
Программная комбинированная иммуносупрессивная терапия проводится больным с подтвержденным диагнозом апластической анемии, которую поставили на основании данных анализа периферической крови, миелограммы и гистологических препаратов костного мозга (билатеральная трепанобиопсия ), и также если отсутствуют противопоказания.
Противопоказаниями же к проведению комбинированной иммуносупрессивной терапии являются тяжелые соматические заболевания, сопровождающиеся сердечно-сосудистой ,почечной, печёночной, дыхательной недостаточностью. Геморрагический синдром инфекционные осложнения рассматривают как временные противопоказания ,которые должны быть купированы до начала иммуносупрессивной терапии АТГ или до проведения спленэктомии. В случае тяжелых инфекционных осложнений (сепсис, пневмония) иммуносупрессивной терапии должна предшествовать интенсивная противоинфекционная терапия , проводимая с учетом возбудителя ( бактерии ,грибы, вирусы ). АТГ или циклоспорин назначаются не ранее, чем через пяти-семи дней после нормализации температуры и исчезновения клинической симптоматики.
Если имеется геморрагический синдром, циклоспорин прописывается вместе с заместительной терапией донорскими тромбоцитами.
Длинное применение глюкокортикоидов (ГКС) при апластической анемии сопровождается развитием тяжелых осложнений (кушингоидный синдром, артериальная гипертензия, стероидный диабет, остеопороз, асептический некроз костей, язвенное поражение слизистой желудочно-кишечного тракта и др.) при отсутствии стойкого терапевтического эффекта.
Длинная гранулоцитопения вместе с использованием ГКС, а в особенности с преднизолоном обычно приводит к развитию тяжелейших инфекционных сложностей, которые требуют интенсивной антибактериальной терапии , создающих неблагоприятные условия для начала иммуносупрессивной терапии и ухудшающих его эффективность.
Вот почему в настоящее время ГКС лучше не пользоваться как стартовой терапии апластической анемии.
При наличии нейтропении и температуры выше 38 тридцати восьми градусов зараженным с апластической анемией прописана антибактериальная терапия. Антибиотики широкого спектра действия, с воздействием на грамотрицательные бактерии, в особенности - на синегнойную палочку, назначают максимальных или субмаксимальных дозах. Используются комбинации цефалоспаринов третьего - четвертого поколения (цефоперазон, цефепим, цефтазидим) с амикацином. Внутримышечное введение антибиотиков у пациентов с нейтропенией и тромбоцитопенией недопустимо. Антигрибковая терапия у больных апластической анемией (амфотерицин B , вориконазол, каспофунгин) устраивается на третий-седьмой день фебрильной нейтропении, сохраняющейся при терапии антибиотиками широкого спектра действия.
Главное место в вылечивании апластической анемии занимает заместительная терапия препаратами крови. Обычно чтобы купировать анемический синдром при апластической анемии используется эритроцитарная масса. Трансфузию донорских тромбоцитов проводят с использованием тромбоконцентрата. При тяжёлом геморрагическом синдроме, развитии ДВС-синдрома показано введение свежезамороженной плазмы.
При трансфузиях эритроцитарной массы срочно нужен контроль показателей обмена железа, в первую очередь ферритина. Содержание ферритина в крови более 1000 нг/мл требует дополнительного назначения дефероксамина (десферала) или деферазирокса (эксиджада).
Комбинированная иммуносупрессивная терапия
1 этап. АТГ устанавливается на первой стадии лечения. Спустя две-три недели от начала курса АТГ (после купирования симптомов сывороточной болезни) начинают терапия циклоспорином А. Начальная доза в сутки ЦсA -10 мг/кг. Коррекция суточной дозы проводится с учетом индивидуальной переносимости препарата и содержания ЦсА в сыворотке крови.
2 этап. Спустя три-шесть месяцев после начала иммуносупрессивной терапии и отсутствие положительной клинико-геологической динамики в программу лечения начинается второй курс терапии АТГ. При нетяжелой апластической анемии может быть проведена спленэктомия.Терапия – ЦсА продолжена.
3 этап. Спустя шесть-двенадцать месяцев начиная с начала иммуносупрессивной терапии при рефлекторном течении апластической анемии и сохраняющиеся зависимости больного от трансфузий донорских эритроцитов и тромбоцитов проводится второй или третий курс АТГ или спленэктомия, если она не была сделана на предшествующих стадиях лечения.
Курс терапии ЦсА у больных апластической анемией продолжается в восемнадцать - двадцать четыре месяца и может быть даже дольше (однако не меньше, чем двенадцать месяцев после достижения ремиссии).
При первых двадцати одного-двадцати восьми днях лечебной терапии зараженный помещен в асептические условия палаты (одноместной). Следует установить подключичный катетер после начала лечения. Пункция подключичной вены должна осуществляться под контролем количества тромбоцитов периферической крови после трансфузии тромбоконцентрата (но не менее шести доз).
Читайте также: