
Экспрессия генов раковых клеток реферат
Обновлено: 05.07.2024
Глущенко Е.С. 1 Белогубов П.В. 1 Антонова А.В. 1 Семенова М.А. 1 Живодерников И.В. 1 Свеколкин В.П. 1 Викторов Д.А. 1 Мастиленко А.В. 1 Столбовская О.В. 1 Саенко Ю.В. 1
Механизмы радиационно-индуцированной клеточной смерти раковых клеток опосредуются изменением экспрессии большого числа генов и являются скоординированным клеточным ответом, ключевую роль в котором играет белок р53. Для изучения роли гена ТР53 в ответе раковых клеток на радиационное воздействие выполнен транскриптомный анализ дифференциально-экспрессирующихся генов в раковой клеточной линии HCT116 с нормальным и мутантным геном ТР53. В ходе эксперимента раковые клетки подвергались радиационному облучению в дозе 4 Гр. Для анализа профилей экспрессии генов использовали гибридизационные ДНК-чипы высокой плотности. В результате кластерного анализа выявлено 3 группы генов с дифференциальной экспрессией. Анализ белковых взаимодействий кластеризованных генов продемонстрировал, что после радиационного облучения экспрессия связанных генов GREB1, CENPT и SREBF1 индуцируется в клетках линии НСТ-116р53+/+ и подавляется в клетках линии НСТ-116р53-/-. Экспрессия гена ZFAND5 может быть связана с устойчивостью клеток к радиационно-индуцированному апоптозу.

1. Coller H.A. Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion / H.A. Coller, C. Grandori, P. Tamayo, T. Colbert, E.S. Lander, R.N. Eisenman, T.R. Golub // Proc. Natl. Acad. Sci. U S A. – 2000. – Vol. 97, № 7. – P. 3260–3265.
2. Enns L. Low-Dose Radiation Hypersensitivity Is Associated With p53-Dependent Apoptosis / L. Enns, K.T Bogen, J. Wizniak, A.D. Murtha, M. Weinfeld // Mol. Cancer Res. – 2004. – Vol. 2. – Р. 557–566.
3. Ghandhi S. Time-series clustering of gene expression in irradiated and bystander fibroblasts: an application of FBPA clustering / S. Ghandhi, A. Sinha, M Markatou, S. Amundson // BMC Genomics. – 2011. – Vol 12. – № 1. – Р. 2.
4. He G. The Protein Zfand5 Binds and Stabilizes mRNAs with AU-rich Elements in Their 3’-Untranslated Regions / G. He, D. Sun, Z. Ou, A. Ding // Journal of Biological Chemistry. – 2012. – Vol. 287. – № 3. – P. 24967–24977.
5. Junttila M.R. Selective activation of p53-mediated tumour suppression in high-grade tumours / M.R. Junttila, A.N. Karnezis, D. Garcia, F. Madriles, R.M. Kortlever, F. Rostker, D.M. Pham, Y. Seo, G.I Evan, C.P Martins // Nature. – 2 010. – Vol. 468. – Р. 567–71.
6. Olivier M. TP53 mutations in human cancers: origins, consequences, and clinical use / M. Olivier, M. Hollstein, P. Hainaut // Cold Spring Harb. Perspect. Biol. – 2010. – Vol. 2, № 1. – P. a001008.
7. Riley T. Transcriptional control of human p53-regulated genes / T. Riley, E. Sontag, P. Chen, A. Levine // Nat. Rev. Mol. Cell Biol. – 2008. – Vol. 9. – № 5. – P. 402–412.
8. Tu Z. Oncogenic Ras Regulates BRIP1 Expression to Induce Dissociation of BRCA1 from Chromatin, Inhibit DNA Repair, and Promote Senescence / Z. Tu, K. Aird, B.G. Bitler, J. Nicodemus, N. Beeharry, B. Xia, T. Yen, R. Zhang // Developmental cell. – 2011. – Vol. 21. – № 6. – P. 1077–1091.
9. Yang J.Y. ATM, ATR and DNA-PK: initiators of the cellular genotoxic stress responses/ J.Y. Yang, H.E. Hamrick, P.J. Duerksen-Hughes // Carcinogenesis. – 2003. – Vol. 24. – № 10. – P. 1571–1580.
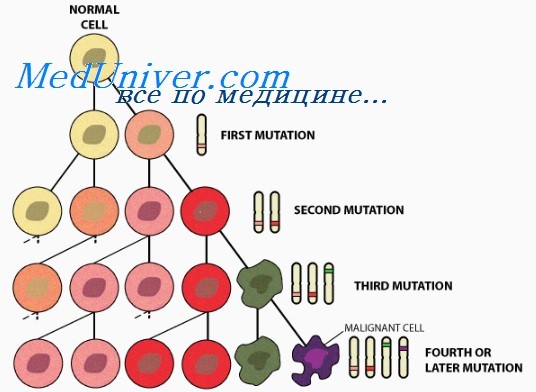
Механизмы радиационно-индуцированной клеточной смерти раковых клеток опосредуются изменением экспрессии большого числа генов и являются скоординированным клеточным ответом, ключевую роль в котором играет белок р53. P53 способен вызвать апоптоз клеток в ответ на повреждение ДНК [5]. Генетический анализ опухолевых клеток человека продемонстрировал ключевую роль р53 в подавлении онкологических процессов. Больше половины опухолей человека из всего широкого спектра типов несут мутации гена TP53, который кодирует белок 53, а наследование мутантного гена ТР53 делает его носителей предрасположенными к онкологическому синдрому Ли–Фраумени [6]. Мутация гена ТР53 затрагивает целый кластер других генов, экспрессия которых зависит от белка р53. Например, запуск p53-зависимого апоптоза связан с индукцией транскрипции компонентов как внешнего, так и внутреннего механизма клеточной смерти, включая такие белки, как BAX, FAS, NOXA и PUMA [7]. Известно, что мутации гена ТР53 приводят к существенным изменениям в механизме клеточного ответа на стрессовые воздействия, в частности, на радиационно-индуцированное увеличение внутриклеточной концентрации АФК. Опухоли с мутантным геном ТР53 обладают высокой радиорезистентностью и способностью к метастазированию, что, как полагают, связано с их генетической нестабильностью [9]. Результатом мутации гена ТР53 может стать разрушение нормальных и возникновение новых сетей экспрессирующихся генов, возникновение новых белковых взаимодействий, приводящих к появлению радиорезистентности.
Использование полно-транскриптомных методов анализа позволяет выявить группы, дифферециально-экспрессирующиеся в разных клеточных линиях в ответ на одно и тоже воздействие. Транскриптомные методы анализа приобретают решающее значение в изучении ответа нормальных и раковых клеток на внешние стрессовые воздействия, в частности, радиационное облучение. Один из способов изучения транскриптома основан на технологии гибридизационных ДНК-чипов высокой плотности. Основным достоинством технологии является возможность одновременного анализа экспрессии всей совокупности экпрессирующихся генов. Это в свою очередь позволяет изучать геном как целостную систему [3].
Целью настоящей работы стал поиск кластеров дифференциально-экспрессирующихся в раковых клетках с нормальным и мутантным геном ТР53 после генов скоординированная экспрессия генов, которых повторяет динамику изменения внутриклеточной концентрации АФК после радиационного облучения в раковых клетках с мутантным и нормальным по геном TP53.
Материал и методы исследования
В экспериментах использовали клеточные линии:
1) клеточная линия рака кишечника HCT-116 p53-/- (мутантная по гену ТР53);
2) клеточная линия рака кишечника HCT-116 p53+/+ (гены ТР53 дикого типа).
Клетки культивировали при 37 °С, во влажной атмосфере, содержащей 5 % СО2. Для культивирования использовали среду RPMI-1649 или DMEM, содержащую L-глутамин, 12 % фетальной коровьей сыворотки и 50 мкг/мл гентамицина. Клетки облучали рентгеновским излучением, генерируемым терапевтическим акселератором Cliniac 600 при комнатной температуре в дозах 4 Гр одноразово. Мощность дозы составляла 0,03 Гр/с, при фокусном расстоянии 104 см. Высота водяного столба над клетками составляла 1 см. Клетки облучались в 24 луночных планшетах (объём лунки 2,5 мл).
Профили экспрессии генов изучались с использованием гибридизационных ДНК-микроматриц высокой плотности серии HGU133A (Human Genome U133A) фирмы Affymetrix (Санта-Клара, Калифорния, США) через 15 мин, 12 и 24 часа после радиационного воздействия. РНК выделялось с использованием набора RNeasy Mini (Qiagen, США). Качество выделенной РНК оценивалось по целостности 18S и 28S рибосомальной РНК с использованием с помощью электрофореза в 1 % агарозном геле. Библиотека клонированных ДНК готовилась с использованием набора GeneChip Expression 3’-Amplification One-Cycle cDNA Synthesis Kit (Affymetrix). Мечение биотином антисмысловых библиотек клонированных РНК и очистка проводилась с использованием набора GeneChip Expression 3’-Amplification Reagents for IVT Labeling (Affymetrix). Матрица окрашивалась стрептовидин-фикоэритрином и сканировалась на сканере GeneAtlas System (Affymetrix, Санта-Клара, Калифорния, США) [1]. Обработка данных полученных после сканирования ДНК-микроматриц, проводилась с использованием алгоритмов MAS5.
Результаты исследования и их обсуждение
На рис. 1 представлены данные сравнительного кластерного анализа динамики транскриптома клеточных линий НСТ-116р53+/+ (с геном ТР53 дикого типа) и НСТ-116р53-/- (с мутированным геном ТР53). Из рис. 1 можно увидеть, что существует несколько кластеров генов динамика экспрессии, которые значительно отличаются между клеточными линиями НСТ-116р53+/+и НСТ-116р53-/-. 1-й кластер представлен 1 геном – RAB2A и PHF14 (группа 1, рис. 1). 2-й кластер генами NME5, SURF2, RRP15, TSFM, RNASEH2B, COPS8, DPH5, ALMS1, FBXO11, MDM1, C5orf54, CCNE2, BRIP1, QRSL1 и ERVMER34-1 (группа 2, рис. 1). 3-я группа генов представлена SREBF1, LTBP4, ZFAND5, GREB1, CENTP и GNAT3 (группа 3, рис. 1). Гены RAB2A и PHF14 в клетках линии НСТ-116р53+/+ имеют сниженную экспрессию по сравнению с контролем на протяжении всего эксперимента (1,12 и 24 ч), тогда как в клетках линии НСТ-116р53-/- их экспрессия значительно увеличена по сравнению с контрольной группой во всех временных точках. Эти гены не связаны между собой, и их продукты не вступают во взаимодействие. Ген RAB2A кодирует белок, относящийся к семейству онкогенов RAS. Его функции заключаются в транспорте комплекса RAS из эндоплазматического ретикулума в аппарат гольджи. Ген PHF14 кодирует белок с неясными функциями. Гены 2 кластера кодируют белки, относящиеся к различным сигнальным и метаболическим путям. Но все они имеют общий профиль экспрессии. В клетках линии НСТ-116р53+/+ с нормальным геном ТР53 через 1 час после радиационного воздействия их экспрессия подавлена в сравнении с контрольной группой. В клетках с мутантным геном ТР53 (линия НСТ-116р53-/-) экспрессия этого кластера генов увеличена, т.е. находится в противофазе по отношению к экспрессии этих же генов в линии НСТ-116р53+/+. Ген BRIP1 кодирует BRCA1, взаимодействующий белок С-концевой хеликазы 1.

Рис. 1. Дендрограмма, отражающая кластеризацию генов, дифференциально экпрессирующихся в клетках линии НСТ-116р53+ (с геном ТР53 дикого типа) и НСТ-116р53- (с мутантным геном ТР53) после облучения гамма-излучением. (Примечание. Чем темнее цвет, тем выше экспрессия гена по сравнению с контрольной группой. Достоверность отличий по отношению к контрольной группе установлена на уровне р
Рак — одна из наиболее частых и серьезных болезней, наблюдаемых в клинической медицине. Статистика показывает, что некоторые формы рака встречаются у более чем одной трети людей, вызывая более 20% всех смертей, и в развитых странах требуют более 10% общих расходов на медицинское обслуживание. Раковые опухоли при отсутствии лечения всегда приводят к летальному исходу.
Ранняя диагностика и раннее лечение жизненно необходимы, и немаловажная цель исследования рака — выявление людей с повышенным риском раковых опухолей до их развития.
В дальнейших статьях на нашем сайте МедУнивер попробуем разобраться, каким образом молекулярно-генетические исследования показывают, что рак — в основном генетическая болезнь. Во-первых, опишем типы генов, вовлеченных в развитие рака, и механизмы, благодаря которым дисфункция этих генов может заканчиваться болезнью.
Во-вторых, рассмотрим множество наследуемых онкологических синдромов и покажем, как понимание их патогенеза высветило основу более частых спорадических форм рака. Мы также изучим некоторые специальные проблемы, возникающие в медицинской генетике и генетическом консультировании в связи с наследуемыми синдромами.
В-третьих, покажем, как генетика и геномика изменили наши представления о причинах рака и методах его диагностики и лечения. Геномика за счет идентификации конкретных делеций и дупликаций сегментов генома раковых клеток и полного анализа экспрессии генов и мутаций в раковых клетках действительно изменила диагностику и лечение рака.
Рак — не одно заболевание, это название используют для обозначения злокачественных новообразований, характеризующихся неконтролируемым клеточным ростом, приводящим к их развитию. Новообразование, чтобы быть раком, должно также быть злокачественным.
Это означает, что его рост больше не контролируется, и опухоль способна прорастать смежные ткани или распространяться (метастазировать) в более отдаленные участки, или и то, и другое одновременно. Опухоли, не способные к прорастанию или метастазированию, не относятся к раковым и называются доброкачественными опухолями, хотя их размер и расположение могут вызывать беспокойство, но в целом они благоприятны для пациента.
Существует три основных формы злокачественных новоообразований: саркомы, когда опухоль возникает в мезенхимальной ткани, например в костях, мышцах, соединительной ткани или в тканях нервной системы; карциномы, возникающие в эпителиальной ткани, скажем, в эпителии клеток кишечника, бронхов или протоках грудной железы; и злокачественные неоплазии гемопоэтической и лимфоидной ткани, например лейкозы и лимфомы, захватывающие костный мозг, лимфатическую систему и периферическую кровь.
В пределах каждой из этих основных групп опухоли классифицируются по их расположению, типу ткани, гистологическим проявлениям и степени злокачественности.
Генетические основы рака
• Независимо от того, появился рак спорадически, в результате соматической мутации или у многих членов одной семьи как наследственный признак, это генетическое заболевание.
• Онкоген — мутантный аллель протоонкогена, класса нормальных генов, кодирующих белки клетки, обеспечивающие рост и выживание клеток. Онкогены облегчают злокачественное перерождение, стимулируя пролиферацию или тормозя апоптоз. Онкогены кодируют такие белки, как:
- белки сигнальных путей пролиферации клеток;
- факторы транскрипции, управляющие экспрессией обеспечивающих рост генов;
- ингибиторы механизмов программируемой смерти клетки.
• Развитие опухоли. После появления рак развивается, накапливая генетические поломки, благодаря мутациям или эпигенетическому подавлению генов ХКЦ, кодирующих механизмы репарации поврежденной ДНК и поддерживающих цитогенетически нормальное состояние. Другое последствие генетических дефектов — изменение экспрессии генов, приводящее к васкуляризации и распространению опухоли инвазивным ростом и метастазированием.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Раковые тестикулярные антигены (РТА) или CancerTestisAntigens (CTA) представляют собой большое семейство опухолевых антигенов, экспрессирующихся, в основном, опухолями человека. Такая топически ограниченная экспрессия вместе с естественными сильными иммуногенными свойствами превращают РТА в идеальную мишень для разработки специфических иммуно-терапевтических подходов. Целью данного обзора было обобщение основных известных механизмов регуляции экспрессии РТА и выполняемых ими функциях, знание которых может быть полезно для создания новых вакцин и оптимизации их работы при лечении онкологических заболеваний. Фармакологическое воздействие на профили экспрессии РТА с помощью ДНК гипометилирующих препаратов обсуждается в качестве возможного подхода для использования в новых схемах комбинированной терапии потенциально способных улучшить эффективность иммунотерапии онкологических пациентов.

1. Водолажский Д.И., Меньшенина А.П., Двадненко К.В. и др. Опыт конструирования дендритно-клеточной вакцины для лечения рака шейки матки // Фундаментальные исследования. – 2015. – № 1. – С. 716-720.
3. Antequera F., Bird A. CpG islands as genomic footprints of promoters that are associated with replication origins // Curr. Biol. 1999. V.9. P.661-667.
4. Boel P., Wildmann C., Sensi M.L., Brasseur R., Renauld J.C., Coulie P., Boon T., van der B.P. BAGE: a new gene encoding an antigen recognized on human melanomas by cytolytic T lymphocytes // Immunity.1995. V.2. P. 167-175.
5. Brasseur F., Rimoldi D., Lienard D. et al. Expression of MAGE genes in primary and metastatic cutaneous melanoma // Int. J. Cancer. 1995. V.63. P. 375-380.
6. Caballero O.L., Chen Y.T. Cancer/testis (CT) antigens: potential targets for immunotherapy // Cancer Sci. 2009. V.100. P. 2014-2021.
7. Calabro L., Fonsatti E., Altomonte M. et al. Methylationregulated expression of cancer testis antigens in primary effusion lymphoma: immunotherapeutic implications // J. Cell Physiol. 2005. V. 202. P. 474-477.
8. Chen Y.T., Scanlan M.J., Sahin U. et al.A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening // Proc. Natl. Acad. Sci. USA.1997. V.94. P. 1914-1918.
9. Chen C.H., Huang G.T., Lee H.S. et al.High frequency of expression of MAGE genes in human hepatocellular carcinoma // Liver. 1999. V.19. P. 110-114.
10. De Smet C., Courtois S.J., Faraoni I. et al.Involvement of two Ets binding sites in the transcriptional activation of the MAGE1 gene // Immunogenetics.1995. V. 42. P. 282-290.
11. De Smet C., De Backer O., Faraoni I. et al.The activation of human gene MAGE-1 in tumor cells is correlated with genome-wide demethylation // Proc. Natl. Acad. Sci. USA.1996. V.93 P. 7149-7153.
12. De Smet C., Lurquin C., Lethe B., Martelange V., Boon T. DNA methylation is the primary silencing mechanism for a set of germ line- and tumor-specific genes with a CpG-rich promoter // Mol. Cell Biol. 1999. V.19. P. 7327-7335.
13. De Smet C., Loriot A., Boon T. Promoter-dependent mechanism leading to selective hypomethylation within the 5’ region of gene MAGE-A1 in tumor cells // Mol. Cell Biol. 2004. V.24. P. 4781-4790.
14. dos Santos N.R., Torensma R., de Vries T.J. et al.Heterogeneous expression of the SSX cancer/testis antigens in human melanoma lesions and cell lines // Cancer Res. 2000. V.60. P. 1654-1662.
15. Fratta E., Sigalotti L., Colizzi F. et al.Epigenetically regulated clonal heritability of CTA expression profiles in human melanoma // J. Cell Physiol. 2010. V.223. P. 352-358.
16. Gattei V., Fonsatti E., Sigalotti L. et al.Epigenetic immunomodulation of hematopoietic malignancies // Semin. Oncol. 2005. V.32. P. 503-510.
17. Gaugler B., van den E.B., van der B.P. et al. Human gene MAGE-3 codes for an antigen recognized on a melanoma by autologous cytolytic T lymphocytes // J. Exp. Med. 1994. V.179. P. 921-930.
18. Gjerstorff M.F., Kock K., Nielsen O., Ditzel H.J. MAGE-A1, GAGE and NY-ESO-1 cancer/testis antigen expression during human gonadal development // Hum Reprod. 2007. V. 22(4). P.953–960.
19. Gure A.O., Chua R., Williamson B. et al. Cancer-testis genes are coordinately expressed and are markers of poor outcome in non-small cell lung cancer // Clin. Cancer Res. 2005. V.11. P. 8055-8062.
21. Jones P.L., Veenstra G.J., Wade P.A. et al.Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription // Nat. Genet. 1998. V.19. P. 187-191.
22. Iizuka M., Smith M.M., Functional consequences of histone modifications // Curr. Opin. Genet. Dev. 2003. V.13. P. 154-160.
23. Jungbluth A.A., Chen Y.T., Stockert E. et al. Immunohistochemical analysis of NY-ESO-1 antigen expression in normal and malignant human tissues // Int. J. Cancer.2001. V.92. P. 856-860.
24. Karpf A.R., Jones D.A. Reactivating the expression of methylation silenced genes in human cancer // Oncogene.2002. V.21. P. 5496-5503.
25. Karpf A.R. A potential role for epigenetic modulatory drugs in the enhancement of cancer/germ-line antigen vaccine efficacy // Epigenetics.2006. V.1. 116e120.
27. Klose R.J., Bird A.P. Genomic DNA methylation: the mark and its mediators // Trends Biochem. Sci. 2006. V.31. P. 89-97.
28. Kobayashi Y., Higashi T., Nouso K. et al.Expression of MAGE, GAGE and BAGE genes in human liver diseases: utility as molecular markers for hepatocellular carcinoma // J. Hepatol. 2000. V.32. P. 612-617.
29. Kurashige T., Noguchi Y., Saika T. et al. Ny-ESO-1 expression and immunogenicity associated with transitional cell carcinoma: correlation with tumor grade // Cancer Res. 2001. V.61. P. 4671-4674.
30. Landry C., Brasseur F., Spagnoli G.C. et al.Monoclonal antibody 57B stains tumor tissues that express gene MAGE-A4 // Int. J. Cancer 2000. V.86. P. 835-841.
31. Li M., Yuan Y.H., Han Y. et al. Expression profile of cancer-testis genes in 121 human colorectal cancer tissue and adjacent normal tissue // Clin. Cancer Res. 2005. V.11. P. 1809-1814.
32. Maio M., Coral S., Fratta E., Altomonte M., Sigalotti L. Epigenetic targets for immune intervention in human malignancies // Oncogene.2003. V. 22. P. 6484-6488.
33. Mashino K., Sadanaga N., Tanaka F. et al. Expression of multiple cancer-testis antigen genes in gastrointestinal and breast carcinomas // Br. J. Cancer.2001. V.85. P. 713-720.
34. Melloni G., Ferreri A.J., Russo V. et al. Prognostic significance of cancer-testis gene expression in resected nonsmall cell lung cancer patients // Oncol. Rep. 2004. V.12.P. 145-151.
35. Rimoldi D., Salvi S., Schultz-Thater E., Spagnoli G.C., Cerottini J.C. Anti-MAGE-3 antibody 57B and anti- MAGE-1 antibody 6C1 can be used to study different proteins of the MAGE-A family // Int. J. Cancer.2000. V.86. P. 749-751.
36. Roeder C., Schuler-Thurner B., Berchtold S. et al. MAGE-A3 is a frequent tumor antigen of metastasized melanoma // Arch. Dermatol. Res. 2005. V.296. P. 314-319.
37. Ruault M., van der Bruggen P., Brun M.E. et al. New BAGE (B melanoma antigen) genes mapping to the juxtacentromeric regions of human chromosomes 13 and 21 have a cancer/testis expression profile // Eur J Hum Genet. 2002. V.10 (12). P. 833-40.
38. Santos-Rosa H., Caldas C. Chromatin modifier enzymes, the histone code and cancer // Eur. J. Cancer.2005. V.41. P. 2381-2402.
39. Sahin U., Tureci O., Chen Y.T. et al. Expression of multiple cancer/testis (CT) antigens in breast cancer and melanoma: basis for polyvalent CT vaccine strategies // Int. J. Cancer.1998. V.78. P. 387-389.
40. Scanlan M.J., Altorki N.K., Gure A.O. et al. Expression of cancertestis antigens in lung cancer: definition of bromodomain testis-specific gene (BRDT) as a new CT gene, CT9 // Cancer Lett. 2000. V.150. P. 155-164.
41. Scanlan M.J., Gure A.O., Jungbluth A.A., Old L.J., Chen Y.T., Cancer/testis antigens: an expanding family of targets for cancer immunotherapy //Immunol. Rev. 2002. V.188 P. 22-32.
42. Scanlan M.J., Simpson A.J., Old L.J. The cancer/testis genes: review, standardization, and commentary // Cancer Immun. Jan.2004. V.23. P. 4-11.
44. Sigalotti L., Coral S., Nardi G. et al. Promoter methylation controls the expression of MAGE2, 3 and 4 genes inhumancutaneous melanoma // Int. J. Cancer.2002. V.25. P. 16-26.
45. Sigalotti L., Fratta E., Coral S. et al. Intratumor heterogeneity of cancer/testis antigens expression in human cutaneous melanoma is methylationregulated and functionally reverted by 5-aza-20-deoxycytidine // Cancer Res. 2004. V.64. P. 9167-9171.
46. Sigalotti L., Fratta E., Coral S. et al. Epigenetic drugs as pleiotropic agents in cancer treatment: biomolecular aspects and clinical applications // J. Cell Physiol. 2007. V.212. P. 330-344.
47. Sigalotti L., Covre A., Zabierowski S. et al. Cancer testis antigens in human melanoma stem cells: expression, distribution, and methylation status // J. Cell Physiol. 2008. V.215. P. 287-291.
48. Simpson A.J., Caballero O.L., Jungbluth A., Chen Y.T., Old L.J. Cancer/testis antigens, gametogenesis and cancer // Nat Rev Cancer. 2005. V. 5(8). P.615–625.
49. Song X., Song W., Wang Y. et al. MicroRNA-874 Functions as a Tumor Suppressor by Targeting Cancer/Testis Antigen HCA587/MAGE-C2 // Journal of Cancer. 2016. V. 7(6). P. 656-663. doi: 10.7150/jca.13674.
50. Tachibana M., Sugimoto K., Nozaki M. et al. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis // Genes Dev. 2002. V.16. P. 1779-1791.
51. Tachibana M., Ueda J., Fukuda M. et al. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9 // Genes Dev. 2005. V.19. P. 815-826.
52. Tajima K., Obata Y., Tamaki H. et al. Expression of cancer/testis (CT) antigens in lung cancer // Lung Cancer.2003. V.42. P. 23-33.
53. Van Der Bruggen P., Zhang Y., Chaux P. et al. Tumor-specific shared antigenic peptides recognized by human T cells //Immunol. Rev. 2002. V.188. P. 51-64.
54. Van der Bruggen P., Traversari C., Chomez P. et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma // Science. 1991.V.13. P. 1643-1647.
55. Wade P.A., Gegonne A., Jones P.L. et al. Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation // Nat. Genet. 1999. V. 23. P. 62-66.
56. Wade P.A. Methyl CpG binding proteins: coupling chromatin architecture to gene regulation // Oncogene.2001. V.20. P. 3166-3173.
57. Watt F.M. Epidermal stem cells: markers, patterning and the control of stem cell fate //Philos Trans R SocLond. B.Biol Sci.1998. V.353. p. 831-837.
58. Weber J., Salgaller M., Samid D. et al. Expression of the MAGE-1 tumor antigen is up-regulated by the demethylating agent 5-aza-20-deoxycytidine // Cancer Res. 1994. V.54. P. 1766-1771.
59. Weinert B.T., Krishnadath K.K., Milano F. et al. Real-time PCR analysis of genes encoding tumor antigens in esophageal tumors and a cancer vaccine // Cancer Immun. 2009. V.9. P. 9.
60. Weiser T.S., Guo Z.S., Ohnmacht G.A. et al. Sequential 5-aza-20-deoxycytidinedepsipeptide FR901228 treatment induces apoptosis preferentially in cancer cells and facilitates their recognition by cytolytic T lymphocytes specific for NY-ESO-1 // J. Immunother. 2001a. V.24. P. 151-161.
61. Weiser T.S., Ohnmacht G.A., Guo Z.S. et al. Induction of MAGE-3 expression in lung and esophageal cancer cells // Ann. Thorac. Surg. 2001b. V.71. P. 295-301.
62. Wischnewski F., Pantel K., Schwarzenbach H. Promoter demethylation and histone acetylation mediate gene expression of MAGE-A1, -A2, -A3, and -A12 in human cancer cells // Mol. Cancer Res. 2006. V.4. P. 339-349.
63. Zhang Y., Ng H.H., Erdjument-Bromage H. et al. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation // Genes Dev. 1999. V.13. P. 1924-1935.
Дополнительные доказательства трофобластной теории рака были получены после открытия белковых антигенов, которые присутствуют только в зародышевых клетках, трофобластах и опухолях. Эти антигены первоначально описали как раковые тестикулярные (РТ) антигены на основании тканевой локализации их паттерна экспрессии (тестикулярная локализация). Однако последние данные позволяют предположить, что эту группу генов более правильно рассматривать как раково-герминальные (РГ) антигены, так как они обычно экспрессируются в эмбриональных яичниках [18]. Обнаружение РГ (или РТ) антигенов подтверждает теорию, согласно которой одной из причин онкогенеза является ненормальная активация генов гамет в соматических клетках.
В последние десятилетия усилия сообщества онко-иммунологов были направлены на поиски тумор-ассоциированных антигенов (ТАА), способных вызвать направленный против опухоли иммунный ответ, и создание соответствующих вакцин. Значительные успехи были сделаны в области идентификации ТАА человека, распознаваемых Т-клетками. В начале 1990-х годов, Бун и др. сообщили о первом успешном клонировании антигена опухоли человека, названного меланомным антигеном-1 (MAGE-1). MAGE-1 вызывал специфический ответ аутологичных цитотоксических Т-лимфоцитов (ЦТЛ) у пациента с меланомой [53].
Раковые тестикулярные антигены (РТА) или Cancer Testis Antigens (CTA) представляют собой большое семейство опухолевых антигенов, экспрессирующихся в опухолях человека различного гистологического происхождения, а в нормальных тканях – только в семенниках и плаценте. Такая топически ограниченная экспрессия вместе с естественными сильными иммуногенными свойствами превращают РТА в идеальную мишень для разработки специфических иммуно-терапевтических подходов при лечении опухолей. Разработано несколько ДКВ-вакцин с использованием РТА, проходящие клинические испытания [1; 2; 64; 65]. В связи с интересом для практического применения в последнее время были предприняты значительные усилия для более детального изучения характеристик биологии РТА. На сегодняшний день идентифицировано 70 семейств РТА, включающих в себя около 140 членов. Большинство из этих РТА экспрессируются в процессе сперматогенеза, но их функции до сих пор неизвестны. По всей видимости, эпигенетические события, особенно метилирование ДНК и пост-транскрипционная регуляция, являются основными механизмами, контролирующими экспрессию РТА в нормальных, онко-трансформированных, а также в раковых стволовых клетках.
Цель данного обзора – общий анализ и предоставление информации об основных механизмах регуляции экспрессии РТА и выполняемых ими функциях, знание которых может быть полезно для создания новых вакцин и оптимизации их работы при лечении онкологических заболеваний. Фармакологическое воздействие на профили экспрессии РТА опухолевыми клетками с помощью ДНК гипометилирующих препаратов обсуждается в качестве возможного подхода для использования в новых схемах комбинированной терапии потенциально способных улучшить эффективность иммунотерапии онкологических пациентов.
1. Экспрессия РТА в нормальных и раковых тканях
В тканях семенников гены Х- РТА экспрессируются, прежде всего, в сперматогониях – пролиферирующих половых клетках, в то время как non-X- РТА экспрессируются на более поздних стадиях дифференцировки, например, в сперматоцитах [48]. В дополнение к экспрессии в тестикулах, экспрессия MAGE-A3, MAGE-A8, MAGE-A10, XAGE-2и XAGE-3 была найдена в плаценте [6]. Показано, что некоторые соматические ткани, такие как поджелудочная железа, печень и селезенка экспрессируют мРНК нескольких РТА. Тем не менее, на основании результатов данных количественной RT-PCR, уровни мРНК генов РТА в соматических тканях составляют, как правило,

Обзор
Малигнизация — один из самых загадочных процессов. Что же на самом деле направляет клетку на тернистый путь перерождения?
Автор
Редакторы



Спонсором приза зрительских симпатий выступила компания BioVitrum.
Гончие еще играют во дворе, но дичи не уйти,
как ни мчится она уже сейчас по лесам.
Франц Кафка
Под малигнизацией понимают приобретение здоровыми клетками черт злокачественности, которые мы подробно рассмотрим ниже. Процесс злокачественного изменения можно уподобить дичи из цитаты Кафки, ведь клетка, однажды встав на этот путь, не сможет вернуться и получить свое клеточное здоровье обратно. Важную роль в понимании основ перерождения клеток и их дальнейшего функционирования сыграла медицина, а следом за ней и молекулярная биология. Но начнем с истоков истории рака.
Часть 1. Биография рака
Первые упоминания о раке встречаются в папирусе Эдвина Смита, датируемом 16 веком до нашей эры [1]. Там же отмечается, что данное заболевание не поддается лечению.
Во времена Гиппократа, около 400 года до нашей эры, появилось специальное обозначение рака — karkinos. Разросшаяся опухоль напомнила Гиппократу краба, окутывающего все вокруг клешнями. Современное название онкологии произошло от греческого слова onkos, которое греки использовали для описания опухолей. Однако врачи того времени не различали доброкачественные и злокачественные новообразования, и karkinos Гиппократа не имеет ничего общего с истинным раком.
Гиппократ выдвинул гуморальную теорию, суть которой состояла в том, что каждый недуг является следствием переизбытка одного из четырех гуморов: крови, слизи, желтой желчи и черной желчи.
Первая половина XX века породила еще одну теорию канцерогенеза, недалеко ушедшую от истины. В 1911 году Пейтон Раус, работая в Рокфеллеровском университете в Нью-Йорке, открыл вирус, способный вызывать опухоли у кур. Ученые по всему свету бросились искать вирусы, ответственные за рак именно у человека, однако ничего не могли найти. В 1974 году в Medical World News вирус рака у человека ставили в один ряд с НЛО, снежным человеком и лохнесским чудовищем. Вирус СВ-40 и вирус папилломы человека, вызывающие рак у людей, были открыты в 1960 и 1983 годах соответственно.
В 1970 году генетик Говард Темин, работавший в лаборатории Макардла в Висконсине и изучавший вирус саркомы Рауса (ВСР, или VSR), представил свою работу на Десятом Международном онкологическом конгрессе. Он открыл у ВСР обратную транскрипцию — синтез ДНК по РНК — и положил начало изучениям ретровирусов. Позднее он отказался от вирусной теории канцерогенеза, а в 1979 году ученые Майкл Бишоп и Харолд Вармус открыли первый протоонкоген — src (сарк), содержащийся в ВСР. Это положило начало новому этапу в истории онкологии, люди наконец-то поняли, как запускается процесс канцерогенеза. Но этого бы не произошло без изучения раковой клетки и ее странной физиологии.
Часть 2. Что заставляет клетку измениться?
В этой главе мы разберем причины злокачественного перерождения клетки. Первым толчком к началу этого изменения является мутация в ДНК.
Важными факторами, вызывающими мутации и провоцирующими раковое перерождение, являются ионизирующее излучение, воздействие ультрафиолетовых лучей, влияние цитотоксических веществ, повреждающих ДНК (к ним относятся наркотические вещества и некоторые лекарственные препараты — например, цисплатин, повреждающий структуру двойной спирали) и органические яды.
Но не всякие повреждения ДНК обязательно приведут к появлению раковой клетки, а лишь те, что затронут определенные гены. Наиболее важную роль в канцерогенезе играют три группы генов: протоонкогены, онкогены и гены — супрессоры опухолей.
Протоонкогены
Основные изменения, происходящие с протоонкогенами:

Рисунок 1. Химерный ген BCR-ABL образуется при слиянии участка 9 хромосомы, несущей ген ABL, с участком 22 хромосомы, несущей ген BCR
В некоторых случаях канцерогенез запускается вирусами. Онкогены в геноме вирусов являются ранее захваченными в клетках-хозяевах нормальными генами, которые со временем превратились в злокачественные. Когда такие онкогенные вирусы попадают в клетку, начинается считывание информации с вирусной ДНК или РНК, в цитоплазме накапливаются онкогенные белки и начинается процесс перерождения.
Онкогены
Онкогены — это гены, активность которых стимулирует образование и развитие злокачественной опухоли. Как уже было упомянуто выше, первый вирусный онкоген был открыт в 1979 году.
Биохимические продукты онкогенов
Онкогены кодируют белки с различной структурой и функциями. К основным продуктам деятельности онкогенов относят:
- Факторы роста. Раковые клетки продуцируют белки, способные вызывать пролиферацию и дифференцировку клеток. Наиболее известным фактором роста является HER2, кодируемый геном ERBB2. Мутации и гиперэкспрессия этого гена обнаружены при раке молочной железы и ассоциированы с крайней агрессивностью опухоли. Суперэкспрессия гена приводит к запуску белковых каскадов, ответственных за клеточное деление. Постоянные сигналы к делению вызывают неконтролируемую пролиферативную активность клеток и их злокачественное перерождение.
- ГТФ-связывающие белки. Гуанозинтрифосфат-связывающие белки участвуют во многих клеточных процессах: передача сигналов, транспорт метаболитов внутри клетки и др. Первыми открытыми ГТФ-связывающими белками были белки семейства Ras — продукты онкогена RAS. При постоянном производстве они вызывают злокачественный рост. Наиболее изученный эффектор Ras — это RAF, который запускает белковый каскад MAPK, отвечающий за клеточное деление и пролиферацию [6].
- Мембранные рецепторы. В онкогенезе основную роль играют рецепторы с тирозинкиназной активностью. Они служат для связывания с ростовыми факторами. К ним относится рецептор эпидермального фактора роста, повышенный синтез которого приводит к перерождению клетки.
- Онкогенные протеинкиназы. Протеинкиназы — это группа ферментов, которые модифицируют белки путем фосфорилирования (присоединения остатка фосфорной кислоты). Протеинкиназы регулируют апоптоз, процессы роста и дифференцировки клеток. Нарушения в их работе приводят к сбою в клеточном цикле и, как следствие, к развитию рака. Например, протеинкиназа AKT1, ответственная за ингибирование апоптоза, при перепроизводстве способна вызывать перерождение клеток. Также, она связана с ростом сосудов в опухоли, что помогает раковым клеткам расселяться по организму и давать метастазы.
Все вышеперечисленные продукты онкогенов являются сигналами к запуску неконтролируемого клеточного деления. Внешние факторы больше не играют никакой роли в жизни клетки, потому что пролиферацию запускают внутренние сигнальные белки.
Гены и белки — супрессоры
В здоровой клетке существуют защитные механизмы, следящие за процессами и регулирующие клеточный цикл. К таким механизмам относят деятельность белков — супрессоров опухолей: p21, p53, pRb, PTEN и др.
Белок p53 — наиболее изученный белок-супрессор. Он является продуктом гена TP53, мутации которого обнаруживаются в клетках многих опухолей [7]. p53 синтезируется во всех клетках организма, но активируется только при повреждениях ДНК. Этот белок способен остановить клеточный цикл и не допускать дальнейшее деление клетки, пока не произойдет репарация ДНК. При сильных повреждениях он также может запускать процесс апоптоза.
Одной из главных функций p53 является сохранение генетической идентичности всех клеток организма. При неправильной работе этого белка клетка получает возможность делиться даже при поврежденной ДНК, что увеличивает вероятность мутаций и накопления дефектных онкогенов. Важную роль в подавлении p53 играет белок MDM2, который в норме регулирует активность p53. Однако при повышенном синтезе он связывается с p53 и ингибирует его противоопухолевое действие.
Эпигенетические факторы рака
Важными факторами канцерогенеза являются эпигенетические события. Эпигенетика изучает процессы, затрагивающие активность генов, но не изменяющие структуру ДНК. К ним относится изменение метилирования ДНК.
Метилирование — это присоединение метильной группы к нуклеотидам в особых, строго определенных участках генома, называемых CpG-островками. Такое изменение не влияет на структуру молекулы, однако может влиять на экспрессию отдельных генов. В частности, если в участке ДНК много метильных групп, то транскрипция этого участка прекращается.
Особенно активно метилирование проходит в эмбриональный период жизни, а у взрослого человека метилировано около 2% генома. В норме баланс между метилированием и деметилированием строго регулируется и соблюдается, однако в старости начинают преобладать процессы метилирования, что может в итоге привести к канцерогенезу. В процессе онкогенеза происходит гиперметилирование CpG-островков, что приводит к общей геномной нестабильности и накоплению еще большего количества мутаций. В большинстве случаев метилированные участки являются промоторами и влияют на активацию или, наоборот, инактивацию генов, что с виду похоже на действие точечных мутаций.
Однако хотя нарушения в эпигенетической регуляции сопровождают развитие злокачественного перерождения, они, как правило, не являются его первопричиной, а лишь одним из сопутствующих факторов.

Рисунок 2. Раковый геном. В процессе жизнедеятельности раковая клетка накапливает огромное количество мутаций и нередко характиризуется полиплоидностью.
Часть 3. Физиологические последствия малигнизации
Основное последствие малигнизации — клеточное бессмертие. Оно может поддерживаться несколькими способами: активацией фермента теломеразы, блокировкой регуляторов митохондриального пути апоптоза и в некоторых случаях активацией механизма ALT (alternative lengthening of telomeres, альтернативного удлинения теломер [14]).
Впервые клеточное бессмертие раковых клеток было продемонстрировано в 1951 году на клеточной линии HeLa, взятой у Генриетты Лакс, вскоре скончавшейся от рака шейки матки (рис. 3) [9].

Как правило, малигнизация сопровождается активацией фермента теломеразы. На концах хромосом находятся короткие повторяющиеся участки ДНК, названные теломерами [10]. После каждого деления теломеры укорачиваются, что в итоге приводит к их полному исчезновению и невозможности продолжать деление. Количество возможных делений для клетки названо пределом Хейфлика. Действие теломеразы заключается в восстановлении теломер и превращении клетки в фактически бессмертную, позволяя ей делиться бесконечно долго. Существуют нормальные клетки, в которых также экспрессируется теломераза. Это клетки, которым надо часто делиться: половые, стволовые и клетки эпителия кишечника. Однако теломераза активна в подавляющем большинстве раковых клеток, что играет важную роль в их жизненном цикле.
С другой стороны, в некоторых злокачественных клетках, наравне с активной теломеразой, существует так называемое альтернативное удлинение теломер, или сокращенно ALT [11]. При ALT происходит гомологичная рекомбинация концевых участков хромосом (рис. 4). В норме рекомбинация происходит в процессе мейоза, однако раковые клетки научились достраивать теломеры, используя теломеры другой хромосомы как матрицу [12].

Важно отметить, что раковое бессмертие контролируется не только теломерами, но и ингибированием путей апоптоза, главным из которых является митохондриальный путь. В норме, из митохондрий в цитоплазму выходят митохондриальные белки и образуют апоптотический комплекс — апоптосому, которая и запускает апоптоз. При неправильной работе регуляторных белков, а к ним относятся белки семейства BCL-2, нарушается выход апоптотических белков, что приводит к сбою в процессе апоптоза. В раковых клетках обнаружены нарушения в работе белков BAX и BAK, а также экспрессия ингибиторов клеточной смерти.
Часть 4. Заключение
Читайте также: