
Аминоацидурия у детей реферат
Обновлено: 04.07.2024
Аминоацидемия — повышенное содержание в сыворотке крови одной или нескольких аминокислот; возникает в результате наследственно обусловленных расстройств обмена аминокислот или нарушения дезаминирующей функции печени. Как правило (но не всегда), аминоацидемия сопровождается аминоацидурией.
Аминоацидурия (аминокислотурия) — выведение повышенного количества аминокислот с мочой или наличие в моче продуктов их обмена, в норме не содержащихся в ней (например, кетоновые тела); развивается преимущественно вследствие наследственных нарушений транспорта аминокислот через эпителий почечных канальцев. Суммарная частота различных урий (включая фенилкетонурию) достигает (по некоторым и скорее заниженным оценкам) 1:200 новорождённых.
• Аминоацидурия двухосновная — наследственный дефект ( r ) транспорта лизина, орнитина и аргинина (но не цистина). Тип 1 (*222690): диарея, выраженное отставание в умственном развитии, непереносимость фенотиазинов. Тип II (222700, 14q11.2, ген LPI) изучен у финнов: для гомозигот характерна гипераммониемия, непереносимость белка (лизинурическая).
• Аргининосукцинатная ацидурия. Аргининосукцинат лиаза — фермент цикла мочевины (*207900, КФ 4.3.2.1, 7cen–q11.2, ген ASL, r ) аргининосукциназа катализирует расщепление N-(l-аргинино)сукцината до l-аргинина и фумарата; при наследуемом нарушении повышена экскреция аргининянтарной кислоты, наблюдаются эпилепсия, атаксия, задержка умственного развития, поражения печени.
• Аспартатглутаматурия (222730, 9p24, ген EAAC1, r ). Клинически: умственная отсталость, гипогликемия голодания.
• Гомоцистинурия — расстройство обмена метионина, характеризующееся выделением гомоцистина с мочой, задержкой умственного развития, эктопией хрусталика, редкими светлыми волосами, вывернутыми наружу коленями, тенденцией к судорожным реакциям, анемией, явлениями тромбоэмболии (ИБС) и жировым перерождением печени. Существует несколько наследственных ( r ) форм заболевания. Распространённость по наиболее частой форме (недостаточность цистатионин синтетазы) — 1 на 100 000 живорождённых детей. Суммарная частота всех форм — значительно больше. Этиология • Недостаточность цистатион(он) b -синтетазы (*236200, КФ 4.2.1.22, 21q22.3, ген CBS, 9 дефектных аллелей, r ) • Дефект метаболизма витамина B12 (277400) • Недостаточность N(5,10)-метилентетрагидрофолат редуктазы (*236250, КФ 1.5.1.20, ген MTHFR) • Избирательная мальабсорбция витамина B12 (261100, синдром Иммурслунд–Грасбека, 10p12.1; MGA1) •• Цистатион(он)- b -синтетазы недостаточность. Клинически: эктопия хрусталика, высокий риск развития ИМ, умственная отсталость, психиатрическая патология, марфаноидное телосложение, остеопороз, возможен панкреатит. Лабораторно: гомоцистинурия, метионинурия •• Метилентетрагидрофолат редуктазы недостаточность. Умеренная умственная отсталость, психическая патология, высокий риск развития ИБС и другой патологии ССС, мышечная слабость, а также значительный риск развития дефектов нервной трубки у потомства. Лабораторно: гомоцистинурия, нормальное содержание метионина в крови •• Недостаточность метилкобаламина (*236270). Развивается зависимая от витамина В12 гомоцистинурия с мегалобластной анемией и тяжёлой умственной отсталостью. Лабораторно: гомоцистинурия, гипометионинемия, содержание в крови фолиевой кислоты и витамина В12 нормальное.
• Иминоглицинурия — наследственный дефект транспорта пролина, гидроксипролина и глицина (*242600, r ). Клинически: атрофия сосудистой оболочки глаза и сетчатки, отставание в умственном развитии. Лабораторно: гидроксипролинурия, гиперглицинурия, пролинурия.
• Кетоацидурия — см. Кетоацидозы у детей, Кетоацидоз диабетический.
• Метилмалоновая ацидурия — наследственное заболевание, характеризующееся задержкой психического и физического развития, наличием в моче метилмалоновой кислоты, метаболическим кетоацидозом. По патогенезу различают недостаточность метилмалонил-КоА мутазы, мевалонат киназы, метилмалонил-КоА рацемазы и дефекты метаболизма кобаламина.
Мевалонат киназа (*251170, КФ 2.7.1.36, 12q24, ген MVK, r ). Клинически: многочисленные дефекты ЦНС, значительное отставание в росте и развитии, анемия, гепатоспленомегалия, катаракта, мевалоновая ацидемия, мевалонатацидурия.
Метилмалонил-КоА мутаза (*251000, ацидурия метилмалоновая, КФ 5.4.99.2, 6p21.2–p12, ген MCM, множество дефектных аллелей, группа комплементации mut; r ). Заболевание: B12-резистентная метилмалоновая ацидурия (от бессимптомной до обусловливающей летальный исход сразу после рождения). Клинически: эпизоды метаболического кетоацидоза, задержка психического и физического развития, различные неврологические проявления, нейтропения, остеопороз. Лабораторно: непостоянная гиперглицинемия, в моче — метилмалоновая кислота и кетоны с длинной углеродной цепью. Витамин В12 для коррекции бесполезен.
Метилмалонил-КоА рацемаза (251120, метилмалонил-КоА эпимераза, КФ 5.1.99.1, r ;). Клинически — см. Мевалонат киназа.
Дефекты метаболизма кобаламина (cbl, см. также раздел Примечания). Различают следующие дефекты синтеза В12-коферментов (аденозин- и метилкобаламины): cbl A, cbl B, cbl C, cbl D, cbl E, cbl F (витамин В12-чувствительные, все r ); недостаточность метилмалонил-КоА мутазы — витамин В12 резистентная (mut) форма метилмалоновой ацидурии.
• Дефект синтеза аденозинкобаламина (cbl A, *251100, точно дефект не установлен). Проявления разной выраженности, с разнообразной клиникой. Возможны кетоацидоз, сопор, кома, спастические парезы, дистония, судорожные припадки, нейтропения, тромбоцитопения, остеопороз, спонтанные переломы, гиперглицинемия.
• Дефект синтеза аденозинкобаламина (cbl B, *251110). Метилмалоновая ацидурия чувствительна к цианокобаламину.
• Дефект освобождения из лизосом (cbl F, 277380, болезнь кобаламина F). Дополнительные признаки: кожная сыпь, макроцитоз.
• Сочетанная недостаточность (cbl C, *277400, r ) метилмалонил-КоА мутазы (251000, КФ 5.4.99.2) и гомоцистеин: метилтетрагидрофолат метилтрансферазы (*156570, КФ 2.1.1.13) — наиболее частая форма дефекта метаболизма кобаламина. Неврология: острый психоз, апатия, сопор, отставание в умственном развитии, шатающаяся походка, подострая дегенерация спинного мозга. Кровь: гемолитическая анемия, мегалобластная анемия. Почки: гематурия, протеинурия, гемолитико-уремический синдром. Разное: пигментная ретинопатия, марфаноидная внешность, ВПС, лёгочное сердце, метаболический ацидоз, гастрит. Лабораторно: гомоцистинемия и гомоцистинурия, цистатионурия, метилмалонилацидурия, мегалобластоз, панцитопения « дефект метаболизма витамина В12 типа I (клинически близкая форма — cbl D, *277410, дефект метаболизма витамина В12 типа II, также именуется cbl G).
• Метилмалоновая ацидемия и гомоцистинурия (cbl D, *277410). Раннее начало с тяжёлыми проявлениями. Лабораторно: гомоцистинемия и гомоцистинурия; цистатионемия и цистатионурия, метионин в крови ниже нормы, мегалобластоз, панцитопения.
• Орнитинурия развивается при мутациях гена орнитин- d -аминотрансферазы (*258870, КФ 2.6.1.13, 10q26, ген OAT, множество дефектных аллелей, r ). При недостаточности фермента (начало в детском возрасте) развиваются поражения сетчатки, приводящие к слепоте в возрасте 30–40 лет. Лабораторно: повышенное содержание орнитина во всех средах организма, орнитинурия, гипераммониемия. При пиридоксин-чувствительной форме содержание орнитина в средах организма уменьшается под влиянием пиридоксаль фосфата.
• Аспартилгликозаминурия развивается при недостаточности гликозиласпарагиназы (*208400, аспартилгликозиламинидаза, N-аспартил b -глюкозаминидаза, КФ 3.5.1.26, 4q32–q33, ген AGU, 12 аллелей, r ). Фермент расщепляет 2-ацетамидо 1 ( b -L аспартамидо)-1,2-дидезоксиглюкозу. Экскреция этого соединения изучена в Финляндии (частота около 1 на 4000, носительство дефектного гена примерно 1:30). Аспартилгликозаминурия сочетается с тяжёлой умственной отсталостью (третья её причина после трисомии 21 и синдрома ломкой Х хромосомы). Клиническая картина: тяжёлое отставание в умственном развитии, одутловатые щёки, грубые черты лица, широкий нос, толстая шея, эмоциональная лабильность, миоклонические судороги, кардиомиопатия, сколиоз, диарея, макроорхидия, ангиокератома. Лабораторно: аспартилгликозаминурия, вакуолизированные лимфоциты. МКБ-10. E72.8 Другие уточнённые нарушения обмена аминокислот
• Цитруллинурия. Недостаточность аргининосукцинат синтетазы (цитруллинемия, *215700, КФ 6.3.4.5, 9q32–q34, ген ASS, r , сложные гетерозиготы) характеризуется высоким содержанием цитруллина в крови, ликворе, моче, отравлением аммиаком. Тяжёлая форма заболевания ( r , тип I, в половине случаев смерть наступает в периоде новорождённости) манифестирует от рождения. Реже встречается позднего начала цитруллинемия (тип II, этиология не очень понятна, активность фермента уменьшена в печени, но не в других органах). Клиническая картина: рвота, диарея, тяжёлая реакция на белки per os, значительное отставание умственного и физического развития, выраженная симптоматика со стороны ЦНС, включая расстройства поведения. Лабораторно: цитруллинурия, цитруллинемия, гипераммониемия. Примечания • Известно не менее 12 копий гена ASS (включая псевдогены) на гаплоидный геном (в т.ч.: 2cen–p25, 3q12–qter, 4q21–qter, 5 (2 локуса), 6, 7, 9p13–q11, 9q11–q22, 11q, 12, Xp22–pter, Xq22–q26, Ycen–q11) • Аминокислота цитруллин названа по имени водяного арбуза Citrullus vulgaris, содержащего много цитруллина).
• Гиперлизинемия (238700) возникает при недостаточности бифункциональной аминоадипиновой семиальдегид синтетазы (КФ 1.5.1.7, ген AASS, r , тетрамер обладает активностью лизин: a -кетоглутарат редуктазы [*238700] и сахаропин дегидрогеназы [268700]). Клинически: умственная отсталость, судорожные припадки, умеренная анемия, вялые мышцы и суставы, возможны эктопия хрусталика и низкорослость. Обследование: изменения электроэнцефалограммы (ЭЭГ), лизинурия, цитруллинурия, гистидинурия. Примечание: при синдроме мальабсорбции лизина (247950, r ) наблюдается такая же клиническая картина.
Лечение проводят индивидуально (в зависимости от конкретного дефекта). Общие принципы: • Диета с пониженным содержанием белка • Обильное питьё • Ощелачивание мочи для предупреждения образования камней • Пеницилламин (в рефрактерных случаях при цистинурии) • Никотинамид перорально (при болезни Хартнапа) • Литотомия (по показаниям).
Профилактика. Серьёзные и нерешённые проблемы — доступная сеть генетического консультирования и тотальный скрининг новорождённых (как и ЭКГ для идентификации врождённых дефектов ССС).
МКБ-10 • E70.8 Другие нарушения обмена ароматических аминокислот • E71.1 Другие виды нарушений обмена аминокислот с разветвленной цепью • E71.2 Нарушения обмена аминокислот с разветвленной цепью неуточнённые • E72 Другие нарушения обмена аминокислот • E72.0 Нарушения транспорта аминокислот • E72.1 Нарушения обмена серосодержащих аминокислот • E72.2 Нарушения обмена цикла мочевины
ПРИЛОЖЕНИЯ
Алкаптонурия (гомогентизинурия) — врождённое нарушение обмена фенилаланина и тирозина (недостаточность гомогентизат-1,2-диоксигеназы [*203500, КФ 1.13.11.5, ген AKU, 3q2, r ]), характеризующееся экскрецией гомогентизиновой кислоты с мочой. Клинически обычно проявляется остеоартритами. Диагностика • При хранении мочи или добавлении щёлочи моча темнеет (результат образования продуктов полимеризации гомогентизиновой кислоты) • Охроноз (например, голубоватая окраска ушей) и артриты. Лечение симптоматическое. МКБ-10 • E70.2 Нарушения обмена тирозина. Примечание. Охроноз — накопление в соединительной ткани гомогентизиновой кислоты с появлением пигментации. Разволокнение структур, кальциноз и утрата прочности; наблюдается при алкаптонурии, отравлениях фенолами.
Гиперпипеколатемия (239400, аллель гена ZWS1?, r ) — пероксисомная болезнь накопления, в крови увеличено содержание (до 4–5 мг%) пипеколатов, промежуточных продуктов метаболизма лизина. Клиническая картина сходна с проявлениями синдрома Целлвегера, но на первый план выступают выраженное отставание в умственном развитии, поражения сетчатки и печени.
Гиперпролинемия типа I (*239500, 22q11.2, дефект гена PRODH, r ) развивается вследствие недостаточности пролин дегидрогеназы (КФ 1.5.99.8) и проявляется аномалиями почек в сочетании с аминоацидурией (пролинурией, гидроксипролинурией и глицинурией); тип II (*239510, r ) развивается вследствие недостаточности d -1-пирролин-5-карбоксилат дегидрогеназы (КФ 1.5.1.1) и проявляется умственной отсталостью, судорогами в сочетании с аминоацидурией.
Глутарацидурия — наследственное ( r ) метаболическое нарушение с прогрессирующим метаболическим ацидозом, дистонией, атетозом, парезами и бульбарными параличами, отставанием в развитии, печёночноклеточной недостаточностью. Характерна энцефалопатия вследствие глиоза и гибели нейронов в базальных ганглиях. Этиология. Типы и гены: • тип I: 231670, GCDH, 19p13.2; • тип IIA: 231680, ETFA, GA2, 15q23 q25; • тип IIB: 130410, ETFB, 19q13.3; • тип IIC: 231675, ETFDH, 4q32 qter. Ферменты: недостаточность глутарил-КоА дегидрогеназы (*231670, КФ 1.3.99.7), недостаточность флавопротеин-убихинон оксидоредуктазы (231675, КФ 1.5.5.1).
Ксантинурия — наследственное нарушение обмена ксантина вследствие недостаточности ксантин дегидрогеназы (КФ 1.1.1.204, *278300, 2p23-p22, ген XDH, тип I, r ) и сочетанной недостаточности ксантин дегидрогеназы и альдегид оксидазы (КФ 1.1.3.22, 1.2.3.1, 602841, 2q33, ген AOX1, тип II [603592]). Клинически: ксантиновые камни в мочевых путях, гидронефроз, пиелонефрит, миопатия. Лабораторно: ксантинурия, низкие уровни мочевой кислоты в сыворотке и моче, отложения кристаллов в скелетных мышцах. При типе I больные метаболизируют аллопуринол, при типе II — нет. МКБ-10 • E75.5 Другие нарушения накопления липидов.
Оксалоз — наследственное заболевание ( r ) с постоянным высоким уровнем экскреции оксалатов с мочой, прогрессирующей двусторонней мочекаменной болезнью (оксалатные камни) и нефрокальцинозом. Этиологически выделено 2 типа оксалоза: недостаточность аланин-глиоксилат аминотрансферазы (тип 1, 259900, 2q36–q37, ген AGT, дефектен также пероксисомный фермент) и более мягкого течения недостаточность глицерат дегидрогеназы (оксалоз типа 2, глицератацидурия, *260000). Внепочечные отложения оксалатов встречаются на более поздних стадиях заболевания. Смерть наступает от ХПН в детстве или подростковом возрасте. Клинически: уролитиаз, нефрокальциноз, ХПН, блокады сердца, сосудистая недостаточность (спазмы, феномен Рейно, окклюзии; перемежающаяся хромота, гангрена), livedo reticularis, патологические переломы костей, сенсорные невропатии (включая слуховую). Проводится пренатальная диагностика обоих вариантов оксалоза (биопсия ворсин хориона, амниотическая жидкость). Терапия препаратами пиридоксина, приводящими к нормализации уровня экскреции оксалатов, достаточно эффективна, желателен подбор минимальных эффективно действующих доз.
Оротацидурия — наследственная болезнь, обусловленная недостаточностью ферментов, катализирующих превращение оротовой кислоты в цитидиловую, характеризуется тяжёлой мегалобластной анемией и отложением в тканях кристаллов оротовой кислоты. Выделено 2 типа оротатацидурии • 1 тип — недостаточность оротидин-5-фосфат пирофосфорилазы и декарбоксилазы оротидин-5-фосфата • 2 тип — изолированная недостаточность декарбоксилазы оротидин-5-фосфата, активность оротидин-5-фосфат пирофосфорилазы повышена. Клинически: мегалобластная анемия, нечувствительная к витамину B12 и фолиевой кислоте, гипохромные микроцитарные эритроциты, резистентные к назначению железа или пиридоксина, гиперэкскреция оротовой кислоты с мочой, нарушение клеточного иммунитета с предрасположением к тяжёлым инфекционным заболеваниям. Лабораторно: оротацидурия, анемия, поддающаяся лечению уридиловой и цитидиловой кислотами. МКБ-10 • E79 Нарушения обмена пуринов и пиримидинов. OMIM. Оротацидурия (258900, 258920).
Научный прогресс в области клинической и молекулярной генетики, биохимии в последние десятилетия предопределил условия, позволившие выделить из ранее не дифференцированных состояний обширную группу “новых” болезней детского возраста, связанных с
 |
Среди разнообразных наследственных болезней обмена термином “органические ацидемии” обозначается большая группа заболеваний, которые проявляются поражением нервной и других систем и характеризуются накоплением органических кислот в биологических жидкостях организма [4].
К первой категории болезней относятся состояния, обусловленные наследственной недостаточностью ферментов, осуществляющих различные этапы преобразования углеродной цепи окси- и кетокислот — производных аминокислот лейцина, изолейцина, валина, лизина, тирозина, гамма-аминомасляной кислоты и некоторых других. В эту группу входят такие заболевания, как лейциноз, тирозинемия 1-го и 3-го типов, глутаровая ацидемия 1-го типа, пропионовая, метилмалоновая, изовалериановая ацидемии, множественный карбоксилазный дефицит и др.
Подавляющее большинство этих болезней проявляется в раннем возрасте, часто в периоде новорожденности. Первые признаки возникают остро, в виде нейродистресс-синдрома (синдрома острых неврологических нарушений) — наблюдаются повышенная возбудимость (или угнетение) функций нервной системы, судороги, расстройства дыхания (апноэ, ацидотическое дыхание или одышка), мышечная гипотония, анорексия, рвота, иногда атетозы, экстрапирамидные расстройства. Эти состояния повторяются в последующие периоды жизни, причем очередные приступы, как правило, связаны с нарушением режима питания или присоединением инфекции. Постепенно обнаруживается задержка нервно-психического, а иногда и физического развития; у некоторых детей отмечается увеличение размеров печени.
В крови в острой фазе болезни определяются признаки декомпенсированного метаболического ацидоза, нередко кетоз и гипогликемия, повышение концентрации аммония, а также анемия, лейкопения. Как в крови, так и в моче в избытке обнаруживаются органические кислоты. Весьма характерно для многих органических ацидемий аминокислотного ряда появление специфического запаха мочи и пота: запах кленового сиропа — при лейцинозе, сыра или потных ног — при изовалериановой ацидемии, вареной капусты — при тирозинемии 1-го типа и др. Однако этот признак может отсутствовать.
Ко второй категории относятся патологические состояния, обусловленные нарушением биоэнергетических процессов (цикл Кребса), тканевого дыхания и окислительного фосфорилирования в митохондриях клеток. Данную группу составляют синдромы Кернса-Сейра, Лея, MELAS (митохондриальная миопатия, энцефалопатия, лактат-ацидоз, инсультоподобные приступы), MERRF (митохондриальная миопатия, энцефалопатия, разорванные красные фибриллы), NARP (нейропатия, атаксия, пигментный ретинит), лактат-ацидоз и другие виды митохондриальной миопатии. Эти заболевания обычно отличаются тяжестью течения, манифестацией преимущественно в детском возрасте, иногда в неонатальном периоде, хотя известны некоторые формы патологии, проявляющиеся у взрослых. Основные клинические симптомы включают: задержку развития, гипотонию, резкую мышечную слабость, беспокойство или сонливость, судороги, дыхательные расстройства (нейродистресс-синдром), атаксию, кардиомиопатию, нарушение сердечного ритма, нередко офтальмоплегию, нистагм, атрофию зрительных нервов, пигментный ретинит, иногда снижение слуха. Общим для всех этих заболеваний признаком является ацидоз, накопление молочной и пировиноградной кислот в крови и моче. Морфологически в мышцах выявляются различные нарушения строения митохондрий, а также своеобразные изменения мышечных волокон (разорванные красные фибриллы).
Третью категорию болезней, связанных с патологией обмена органических кислот, составляют заболевания, вызванные нарушением транспорта или митохондриального окисления жирных кислот. В эту группу входят: системный дефицит карнитина, дефицит карнитин пальмитоил трансферазы, дефицит длинно- средне- и короткоцепочечных ацил-КоА дегидрогеназ, дефицит 3-гидрокси-3-метилглутарил-КоА лиазы и др. Большинство заболеваний проявляется в раннем детском возрасте, однако некоторые формы нарушенного транспорта жирных кислот отличаются поздним началом и относительно доброкачественным течением.
Среди характерных для данной группы болезней симптомов можно выделить: упорную рвоту, мышечную слабость и гипотонию, нерезко выраженное и непостоянное увеличение печени, при некоторых формах болезни кардиомиопатию, эпизоды мышечных болей и миоглобинурии. Приступы обычно провоцируются длительным голоданием (более 12 часов) или физической нагрузкой.
К специфическим особенностям этого вида патологии относится сочетание гипогликемии с гипокетонемией, аномальный состав органических кислот мочи (повышенное выведение жирных кислот с различной длиной углеродной цепи), снижение уровня свободного карнитина в плазме при увеличении содержания его эстерифицированных форм. Морфологическое исследование выявляет липидные включения в мышечной ткани, жировую инфильтрацию печени. Представленные заболевания являются одной из причин синдрома внезапной младенческой смерти и синдрома Рейе.
Две последние категории болезней, по существу, представляют собой единую группу — митохондриальную патологию, которая сопровождается органической ацидурией.
По способам наследственной передачи среди митохондриальных болезней выделяют [11]:
- заболевания, наследуемые моногенно по менделевскому типу, при которых в связи с мутацией ядерных генов нарушаются структура и функционирование митохондриальных белков или изменяется экспрессия митохондриальной ДНК;
- болезни, вызываемые мутациями митохондриальных генов, которые в основном передаются потомству по материнской линии.
Особое место среди органических ацидемий занимают патологические состояния нарушенного пероксисомного окисления жирных кислот с очень длинной углеродной цепью (С 22 и выше). Среди пероксисомных болезней выделяют три группы в зависимости от степени функциональных и морфологических нарушений пероксисом [9,10]:
- с общей дисфункцией пероксисом; при этом морфологически в тканях пероксисомы практически отсутствуют или их количество и размеры существенно уменьшены (цереброгепаторенальный синдром Цельвегера, неонатальная адренолейкодистрофия, младенческий тип болезни Рефсума и др.);
- с нарушением активности нескольких пероксисомальных ферментов при морфологически не измененных пероксисомах (синдром псевдо-Цельвегера, гиперпипеколовая ацидемия и др.);
- с нарушением активности одного определенного фермента и нормальным количеством пероксисом (взрослый тип болезни Рефсума, Х-сцепленная адренолейкодистрофия, псевдоадренолейкодистрофия и др.).
Пероксисомные болезни могут проявляться в раннем детском возрасте, в том числе в периоде новорожденности (цереброгепаторенальный синдром Цельвегера, синдром псевдо-Цельвегера, младенческий тип болезни Рефсума, неонатальная адренолейкодистрофия). Ряд заболеваний манифестирует в более позднем подростковом периоде (взрослый тип болезни Рефсума, Х-сцепленная адренолейкодистрофия). Пероксисомная патология в основном отличается тяжестью поражения нервной системы, задержкой развития, судорогами, мышечной гипотонией, черепно-лицевой дизморфией, увеличением печени. Некоторым формам свойственна диспропорция скелета с укорочением проксимальных отделов конечностей. Характерно снижение зрения (катаракта, пигментный ретинит) и слуха. Среди биохимических изменений обращает внимание органическая ацидурия в виде повышенной экскреции жирных кислот с большим числом атомов углерода, гипохолестеринемия, повышенная экскреция пипеколовой кислоты, а также дефицит синтеза плазмалогенов, нарушение обмена фитановой кислоты, увеличение в сыворотке уровня промежуточных метаболитов желчных кислот.
Анализ клинических и биохимических проявлений органических ацидемий различного происхождения убеждает в их значительном фенотипическом сходстве. Первые признаки нарушенного состояния у детей появляются, как правило, в раннем возрасте. К ним, за редкими исключениями, относятся: прогрессирующее течение болезни, нарушение физического, статико-моторного развития и умственная отсталость, судороги, гипотония мышц, повышенная возбудимость или угнетение центральной нервной системы, рвота, респираторные расстройства.
Таблица 1. Основные клинические и биохимические признаки органических ацидемий различной природы
В то же время отмечается клинико-биохимическое своеобразие каждой группы органических ацидемий (табл. 1). Так, для ряда заболеваний, обусловленных дисфункцией митохондрий, характерна поздняя манифестация в подростковом возрасте или у взрослых. Обращает внимание, что некоторые клинические и обменные нарушения свойственны только определенной группе болезней. Например, кардиомиопатия, мышечные боли и миоглобинурия часто сопровождают нарушения транспорта и окисления жирных кислот. Необычный запах мочи встречается при заболеваниях, связанных с расстройствами обмена аминокислот и жирных кислот. Однако для аминоацидопатий характерны кетотические состояния, тогда как нарушения окисления жирных кислот проявляются гипокетонемией. Пероксисомные болезни отличаются сочетанием черепно-лицевых микроаномалий с тяжелой умственной отсталостью, диспропорцией скелета, выраженной гепатомегалией, снижением зрения и слуха, гипохолестеринемией.
 |
Таким образом, на основании клинических данных и результатов биохимических исследований в большинстве случаев можно заподозрить у больного патологию обмена органических кислот, в том числе митохондриальные и пероксисомные нарушения определенной природы. Установление точного диагноза требует анализа состава органических кислот крови и мочи с выявлением конкретных метаболитов, накапливающихся в биологических жидкостях. При этом предпочтительнее использование такого информативного метода, как газо-жидкостная хроматография в сочетании с хромато-масс-спектрометрией [6]. При органических ацидемиях аминокислотной природы в моче обнаруживаются дериваты соответствующих аминокислот; для заболеваний, связанных с нарушением энергетического обмена и дисфункцией митохондрий, свойственны повышенная экскреция молочной и пировиноградной кислот, ряда дикарбоновых кислот и др.; для пероксисомных болезней характерно накопление в моче жирных кислот с очень длинной углеродной цепью.
Накопленный опыт позволяет считать, что обследованию на органические ацидемии в первую очередь подлежат дети, страдающие симптомокомплексами, характеризуемыми как нейродистресс- и респираторный дистресс-синдромы. Схема обследования таких детей должна включать:
- генеалогический анализ с изучением родословной;
- оценку физического развития;
- анализ психоречевого и моторного развития;
- исследование состояния внутренних органов (включая кардиологическое обследование и др.);
- оценку неврологического статуса с использованием данных электроэнцефалографии, эхоэнцефалоскопии и электронейромиографии (по показаниям);
- офтальмологическое обследование и др. Этапы биохимической программы идентификации патологии составляют:
- определение кислотно-основного равновесия, уровня молочной и пировиноградной кислот в крови, аминокислотного состава крови и мочи;
- исследование содержания глюкозы, аммиака, холестерина в крови; выявление кетоновых тел, миоглобина в моче;
- определение спектра органических кислот крови и мочи (хромато-масс-спектрометрия, газо-жидкостная хроматография) [1].
При подозрении на болезни митохондриальной и пероксисомной природы показано морфологическое исследование клеток печени, скелетных мышц и лимфоцитов крови с использованием электронно-оптических и гистохимических методов оценки активности митохондриальных ферментов (НАДФ-Н редуктазы, цитохром С оксидазы, АТФ-азы, сукцинатдегидрогеназы) и ферментов пероксисом (5).
Применение в клиниках института разработанной диагностической программы позволило выявить у детей целый ряд заболеваний, связанных с патологией обмена органических кислот (лейциноз, тирозинемия, изовалериановая ацидемия и др.), в том числе болезни, обусловленные нарушением функции митохондрий (синдром Кернса-Сейра, различные виды митохондриальной миопатии, дефицит ацил-КоА дегидрогеназы среднецепочечных жирных кислот и др.) и пероксисомные болезни (ризомелическая точечная хондродисплазия).
Своевременная диагностика заболеваний позволяет осуществлять целенаправленные лечебные мероприятия, способствующие предупреждению поражения центральной нервной и других систем организма, а нередко и устраняющие угрозу жизни пациентов. При большинстве органических ацидемий аминокислотного ряда используется диетотерапия с резким ограничением белка в рационе до 1–1,5 г/сутки. Тиамин-зависимая форма лейциноза поддается лечению большими дозами витамина В1 (10 мг/сутки и выше). Для лечения множественного дефицита карбоксилаз успешно применяется биотин (5-40 мг/сутки). В последнее время в терапии метилмалоновой и пропионовой ацидемий нашли применение антибиотики, в частности ампициллин. Для выведения больных из угрожающих жизни состояний применяются методы диализа.
Терапия заболеваний, связанных с дисфункцией митохондрий, представляет большие трудности. Исключение составляют патологические состояния, обусловленные нарушением транспорта и метаболизма жирных кислот. При лечении болезней этой группы используются диетические мероприятия (дробное питание с небольшими промежутками между приемами пищи, ограничение жиров и обогащение рациона углеводами), карнитин, исключение переохлаждения, физических и эмоциональных перегрузок. Основной принцип терапии других форм митохондриальной патологии — применение кофакторных препаратов: коэнзима Q 30-300 мг/сутки; аскорбиновой кислоты 1000-4000 мг/сутки; рибофлавина 100 мг/сутки; тиамина 200-1000 мг/сутки; карнитина 100 мг/кг/сутки [7].
Способы лечения пероксисомных болезней в настоящее время не отработаны. Отмечены положительные результаты диетотерапии с ограничением жиров и дополнительным введением эфиров липидов, эруковой и олеиновой кислот при Х-сцепленной адренолейкодистрофии и болезни Рефсума. В ряде случаев улучшению состояния больных способствовало использование плазмофереза.
Пути профилактики органических ацидемий связаны с установлением природы болезни, медико-генетическим консультированием семьи, дальнейшей разработкой и внедрением способов пренатальной диагностики заболеваний, основанных при большинстве форм патологии на определении уровня соответствующих органических кислот в амниотической жидкости или оценке активности ключевого фермента в клетках хориона.
Литература
1. Антошечкин А. Г., Максимова Л. А., Ченцова Т. В., Николаева Е. А. Диагностика аминоацидопатий и органических ацидурий. II Всесоюз. съезд мед. генетиков. Алма-Ата, 1990, с. 232.
2. Вельтищев Ю. Е., Казанцева Л. З., Семячкина А. Н. Наследственные болезни обмена веществ. В кн Наследственная патология человека П/ред. Вельтищев Ю. Е., Бочков Н. П. М. 1992, т. 1, с. 41-101.
3. Вельтищев Ю. Е. Состояние здоровья детей и общая стратегия профилактики болезней. Росс.вестник перинатологии и педиатрии. Приложение. М., 1994.
4. Казанцева Л. З., Антошечкин А. Г., Николаева Е. А. и др. Клинико-биохимическая диагностика наследственных форм органических ацидемий у детей. Материнство и детство, 1992, № 2-3, с. 21-25.
5. Николаева Е. А., Казанцева Л. З., Клембовский А. И. и др. Критерии диагностики митохондриальной патологии у детей. Первый (третий) Рос. съезд мед. генетиков. Тез. док. М., 1994, с. 44-45.
6. Chalmers R. A., Lawson A. M. Organic acids in man. London-New York, 1982.
7. Clarke L. A. Mitochondrial disorders in pediatrics. Clinical, biochemical and genetic implications. Pediatr. Clin. North Amer, 1992, vol. 39, P. 319-334.
8. Poulton J. Mitochondrial DNA and genetic disease. Arch. Dis. Childh., 1988, vol. 63, P. 883-885.
9. Schutgens R. B. H., Wanders R. J. A., Niyenhuis A. et al. Genetic diseases caused by peroxisomal dysfunction. Enzyme, 1987, vol. 38, P. 161-176.
10. Stephenson J. B. P. Inherited peroxysomal disorders involving the nervous system. Arch. Dis. Childh., 1988, vol. 63, P. 767-770.
11. Zeviani M., Bresolin N., Gellera C. et al. Nucleus-driven multiple large-scale deletions of the human mitochondrial genome: a new autosomal dominant disease. Am. J. Hum. Genet., 1990, vol. 47, P. 904-914.
Из биохимии
Органическими или жирными кислотами принято называть все углеводородные соединения, имеющие в своей структуре карбоксильную группировку -COOH. При наличии в молекуле аминогруппы жирные кислоты называются аминокислотами; гидроксильная группировка (или кетогруппа) позволяет обозначать их как окси- и кетокислоты. Органические кислоты образуются при обмене аминокислот, углеводов, липидов, а также в процессах тканевого дыхания и транспорта электронов в дыхательной цепи [6]. Соответственно среди органических ацидемий можно выделить различные типы нарушений метаболизма.
Митохондрии — внутриклеточные органеллы, которые играют ключевую роль в энергетическом обмене. Согласно недавно полученным сведениям, митохондрии — единственный источник экстраядерной ДНК у человека. Каждая органелла содержит несколько кольцевидных хромосом. Их гены кодируют отдельные субъединицы комплексов электронно-транспортной цепи (в частности, комплексов 1, 3, 4, 5), а также контролируют синтез группы транспортных и рибосомальных РНК [7].
Пероксисомы представляют собой субклеточные органеллы, встречающиеся во всех тканях, но особенно в больших количествах — в гепатоцитах, клетках проксимальных почечных канальцев, корковом слое надпочечников, миелиновых структурах и жировой ткани. Они активно участвуют в процессах перекисного окисления, b-окислении жирных кислот с очень длинной углеродной цепью, биосинтезе желчных кислот, плазмалогенов (эфиросодержащих фосфолипидов) холестерина и др.
Болезнь Хартнупа – это редкое наследственное заболевание, характеризующееся нарушением обмена аминокислот, главным образом, триптофана. Основные симптомы – пеллагроподобные кожные высыпания на лице и открытых участках тела, фоточувствительность, неврологические расстройства. Диагноз ставится при обнаружении повышенной концентрации в моче нейтральных аминокислот и продуктов их распада, подтверждается молекулярно-генетическим исследованием. Лечебные мероприятия включают изменения в диете, предохранение кожных покровов от солнечных лучей, назначение препаратов никотиновой кислоты.
МКБ-10
Общие сведения
Болезнь Хартнупа (аминоацидурия дикарбоновых кислот) названа в честь первого больного, у которого она была диагностирована. Заболевание впервые было описано английским врачом Д. Бароном в 1956 году. Клинические проявления напоминают картину пеллагры − патологического состояния, вызванного недостаточностью никотиновой кислоты. По различным статистическим данным, распространенность болезни Хартнупа составляет от 1:18 000 до 1:54 000 человек. Значимые гендерные различия отсутствуют.
Причины
Развитие болезни Хартнупа связано с генетической мутацией гена SLC6A19, расположенного в локусе 5p15.33 5 хромосомы. Этот ген кодирует натрий-зависимый белок-переносчик, участвующий в транспорте нейтральных аминокислот (триптофана, тирозина, гистидина и т.д.). Данный белок преимущественно локализован в эпителии тонкого кишечника и почечных канальцев. Наследование заболевания происходит по аутосомно-рецессивному типу. Родители больного ребенка являются здоровыми носителями мутантного гена.
Патогенез
Основу для запуска патологических проявлений составляет дисбаланс в метаболизме триптофана. Из-за нарушения всасывания триптофан задерживается в кишечнике, где под влиянием бактериальной микрофлоры превращаются в продукты распада – индол, кинуренин. Эти соединения, попадая в системный кровоток, отказывают токсическое влияние на центральную нервную систему. Их инактивация происходит в печени, где они соединяются с серной кислотой и выводятся с мочой в виде конъюгатов.
Нарушение реабсорбции аминокислот в канальцах почек ведет к дополнительной их потере с мочой. Триптофан является основным субстратом для синтеза никотиновой кислоты (витамина PP), входящего в состав ферментов, участвующих в тканевом дыхании и других процессах энергетического обмена. Также из триптофана образуется мелатонин – основной гормон, регулирующий циклы сна и бодрствования.
Симптомы болезни Хартнупа
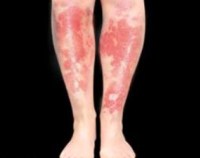
Заболевание имеет хроническое течение с периодами рецидивов и ремиссий. Первые клинические признаки могут возникнуть в любом возрасте. Манифестации и обострению способствуют недостаточное питание, эмоциональный или физический стресс, прием лекарственных препаратов и пр. Самым ярким симптомом болезни Хартнупа является окрашивание мочи в голубой или синий цвет. Это объясняется наличием в моче окисленных продуктов обезвреженного печенью индола.
Наиболее сильно страдают кожные покровы и нервная система. На открытых участках тела (лицо, шея, руки) после длительной инсоляции кожа покрывается красными пятнами, становится грубой, появляются пузыри с серозным содержимым. Субъективно пациент испытывает зуд и жжение в местах высыпаний. Через несколько дней кожа в пораженных областях приобретает коричневатый оттенок, начинает шелушиться, истончаться.
Самыми частыми неспецифическими неврологическими симптомами считаются головная боль, головокружения, бессонница. Часто случаются обмороки. При тяжелом течении наблюдаются нарушение координации движений, неустойчивость при ходьбе, тремор рук и головы. Часто встречаются отклонения в психической сфере, например, постоянные перепады настроения, плаксивость. Очень редки косоглазие, непроизвольные колебательные движения глаз.
Осложнения
Какие-либо серьезные неблагоприятные последствия, представляющие непосредственную угрозу для жизни пациента, страдающего болезнью Хартнупа, крайне редки. Лишь при очень тяжелом течении патологии в единичных случаях у детей младшего возраста возможно глубокое поражение центральной нервной системы, которое приводит к летальному исходу. Также у детей иногда наблюдается отставание в умственном развитии.
Встречаются осложнения со стороны психической сферы – депрессия, острые психозы со зрительными, слуховыми галлюцинациями. После вскрытия пузыри на кожных покровах оставляют язвенные поверхности, что создает благоприятные условия для присоединения вторичной бактериальной флоры (стафилококки, стрептококки). Частые падения вследствие нарушения равновесия и потерь сознания повышают риск черепно-мозговых травм и различных переломов.
Диагностика
Пациентов с болезнью Хартнупа курируют врачи-педиатры, неврологи. В установлении диагноза большую помощь оказывают анамнестические данные (наличие близкого родственника с подтвержденной болезнью Хартнупа). При расспросе больного уточняют, что предшествовало возникновению симптомов (нарушение диеты, инфекционное заболевание, стресс).
При неврологическом осмотре больной демонстрирует затруднения при выполнении координационных тестов – позы Ромберга, пяточно-коленной, пальце-носовой пробы. Чтобы подтвердить диагноз, назначаются следующие дополнительные методы обследования:
- Определение экскреции аминокислот. В анализе мочи выявляются высокие концентрации нейтральных аминокислот, продуктов их распада (индол, индикан). Также отмечается увеличение содержания в моче 5-гидроксииндолуксусной кислоты после пероральной нагрузки триптофаном.
- Биопсия кожи. При гистологическом исследовании биоптата пораженного участка кожи обнаруживаются неспецифические изменения, такие как гиперкератоз, атрофия эпидермиса, умеренная лимфоцитарная инфильтрация.
- Молекулярно-генетический анализ. Методом полимеразной цепной реакции в крови определяется генетическая мутация SLC6A19.
Дифференциальная диагностика
Для исключения других видов аминоацидурий, например, синдрома Фанкони выполняетя анализ мочи на гидроксипролин, аргинин. Данные показатели при болезни Хартнупа остаются нормальными. Также дифференциальный диагноз проводится с дерматологическими заболеваниями (экземой, атопическим дерматитом), наследственной атаксией-телеангиэктазией.
Лечение болезни Хартнупа
В зависимости от тяжести состояния больного лечение может проводиться как амбулаторно, так и в условиях стационара. Пациенту рекомендуется избегать факторов, способствующих возникновению обострения болезни, особенно это касается инсоляции; употреблять достаточное количество белка и витамина гр. В. Защитные мероприятия включают ношение закрытой одежды, использование солнцезащитных кремов в летнее время. Для фармакотерапии болезни Хартнупа применяются:
- Никотиновая кислота. Во время приступа назначается парентеральное введение больших доз никотиновой кислоты, что позволяет добиться быстрого регресса неврологической и дерматологической симптоматики. В дальнейшем пациент должен принимать препараты никотиновой кислоты в пероральной форме.
- Аминокислоты. Тяжелым больным для быстрого восстановления уровня триптофана в крови может понадобиться инфузионное введение аминокислотных смесей, содержащих высокое количество триптофана.
- Глюкокортикостероиды. При выраженном кожном поражении, для снятия воспаления применяются мази или кремы, содержащие ГКС (преднизолон, триамцинолон).
- Психотропные средства. Если развились психические расстройства, используют нейролептики (аминазин), антидепрессанты (флуоксетин), транквилизаторы (диазепам).
Прогноз и профилактика
В подавляющем большинстве случаев болезнь Хартнупа имеет доброкачественное течение и благоприятный прогноз. Продолжительность жизни пациентов при своевременной диагностике и назначении лечения практически не отличается от общей популяции. Известны единичные ситуации с летальным исходом вследствие генерализованного поражения нервной системы.
Методов первичной профилактики не существует. Сбалансированная диета с высоким содержанием белка и витамина PP способствует предотвращению приступов. Если в семье есть родственники с диагностированной болезнью Хартнупа, рекомендуется сделать анализ мочи для определения концентрации аминокислот и пройти медико-генетическое консультирование для раннего выявления заболевания.
Информация только для специалистов в сфере медицины, фармации и здравоохранения!

Авторы: В.П.ЗЫКОВ 1 , д.м.н., профессор, А.Н.ЗАВАДЕНКО 1 , О.А.МИЛОВАНОВА 1 , к.м.н., доцент, И.Л.СТЕПАНИЩЕВ 1,2 , к.м.н., М.Г.САМИГУЛИНА 2
1 – РМАПО, Москва; 2 – ДГКБ №9, Москва
Недостаточность биотинидазы (НБ) –наследственная болезнь обмена из группы органических ацидурий с аутосомно-рецессивным типом наследования. Заболевание обусловлено мутациями в гене, кодирующем фермент биотинидазу, что приводит к нарушению работы всех биотинзависимых карбоксилаз.
Сноска 1. В основе заболевания лежит биотин-дефицитное состояние с развитием недостаточности карбоксилаз.
Распространенность. Заболевание является панэтническим, в европейских странах его частота составляет 1:40000 живых новорожденных [2]. В 14 странах (США, Италия, Испания, Австрия и др.) осуществлен пробный скрининг новорожденных, продемонстрировавший высокую частоту патологии 1:35 000 – 1:54 000, что позволило сделать вывод о необходимости включения данного заболевания в программу массового скрининга. Для этой цели разработаны упрощенные методы определения биотинидазы в высушенных на бумаге образцах крови [3]. В настоящее время скрининг недостаточности биотинидазы проводится в 22 штатах США, многих странах Европы и Японии [18].
В ближайшейперспективе рассматривается возможность проведения в России неонатального скрининга еще на три заболевания: серповидно-клеточную анемию, нарушения обмена жирных кислот и недостаточность биотинидазы. Селективный скрининг на выявление дефицита биотинидазы был впервые проведен в нашей стране еще в 1989-1990 гг. среди больных с патологией нервной системы, кожи, алопецией; частота дефицита биотинидазы в этой группе обследованных составила 1:1000 [3]. В связи с достаточно высокой распространенностью дефицита биотинидазы, ее курабельностью и включением в перспективе в программу скрининга новорожденных, знания о данной патологии необходимы широкому кругу специалистов.
Этиология и патогенез. Ген биотинидазы картирован в сегменте p25 хромосомы 3 [5]. При ДНК-диагностике наиболее часто выявляют мутации G98d7i3 и R538C, встречающиеся более чем в 50% мутантных аллелей [11]. Наиболее частой мутацией при глубокой недостаточности биотинидазы является Q456H [12]; при парциальной недостаточности – D444H [17]. Тип наследования – аутосомно-рецессивный.
В основе заболевания лежит биотин-дефицитное состояние с развитием недостаточности карбоксилаз. Молекула биотина состоит из гетероциклического кольца с прикрепленными к нему карбоксильными группами. Биотин функционирует как коэнзим для 4 карбоксилаз [22]:
- пируваткарбоксилазы (являющейся одним из ключевых ферментов глюконеогенеза);
- пропионил-КоА-карбоксилазы (участвующей в метаболизме аминокислот с разветвленной цепью и некоторых жирных кислот);
- β-метилкротонил-КоА-карбоксилазы (участвующей в метаболизме лейцина);
- ацетил-КоА-карбоксилазы (катализирующей первую стадию биосинтеза жирных кислот).
Фермент биотинидаза участвует в обмене биотина:
- высвобождает биотин, поступающий с пищей и связанный с белком, делает его пригодным для связывания с апокарбоксилазой и образования активного фермента;
- расщепляет биоцитин (биотиниллизин), являющийся продуктом деградации биотинзависимых карбоксилаз, обеспечивая, таким образом, восстановление неустойчивого свободного биотина.
Дефицит биотинидазы приводит к дефициту внутриклеточного биотина, который может быть парциальным (10-30% от средней нормальной активности в сыворотке) или глубоким (менее 10% нормальной активности в сыворотке). Оцениваемая частота обеих форм составляет 1:61000 [22]. Нарушение функции карбоксилаз приводит к накоплению субстратов контролируемых карбоксилазами ферментных реакций. Данные субстраты и их производные токсичны для организма человека и приводят к развитию метаболического ацидоза и вторичной гипераммониемии. Кетоацидоз является признаком продолжительной недостаточности биотина в организме и может не выявляться на начальных этапах болезни [7].
При дефекте биотинидазы особенно уязвима центральная нервная система, т.к. активность биотинидазы в головном мозге человека крайне низкая, и для нормального функционирования нейронов необходимо достаточное и постоянное поступление биотина через гематоэнцефалический барьер. Это является причиной неврологических нарушений, которые в течение определенного периода могут быть единственным признаком заболевания [15]. Описаны изменения в мозжечке, подкорковых структурах, спинном мозге, которые объясняются токсическим эффектом [4,14]. Миелиновые волокна поражаются в большей степени, чем тела нейронов, поэтому при недостаточности биотинидазы часто наблюдается дезорганизация миелиновых структур головного мозга. Причиной изменений кожных покровов в виде алопеции и кожной сыпи является снижение уровня протективных жирных кислот (в результате нарушения работы пропионил-КоА-карбоксилазы) [16]. Развитие нейросенсорной тугоухости может быть связано с накоплением органических кислот, биоцитина, более крупных биотинильных белков [22].
Клинические проявления недостаточности биотинидазы. Тяжесть клинических проявлений зависит от уровня активности биотинидазы. В клинической картине доминируют неврологическая симптоматика, кожные проявления, снижение слуха и зрения, а также вторичные инфекционные осложнения, обусловленные нарушениями иммунитета.
Сноска 2. В клинической картине заболевания доминируют неврологическая симптоматика, кожные проявления, снижение слуха и зрения.
Возраст дебюта составляет от 1 до 6 месяцев. В редких случаях заболевание развивается на первых неделях жизни или в подростковом возрасте [23]. Первыми проявлениями являются мышечная гипотония и судороги. Судороги отмечаются более чем у половины пациентов и представлены генерализованными тонико-клоническими, миоклоническими и инфантильными спазмами. Как мышечная гипотония, так и судороги не снимаются при проведении общепринятой терапии, но достаточно быстро купируются при назначении биотина.
Сноска 3. Возраст дебюта составляет от 1 до 6 месяцев, в редких случаях заболевание развивается на первых неделях жизни или в подростковом возрасте.
К другим симптомам относятся дыхательные расстройства (одышка, тахипноэ/апноэ, ларингеальный стридор), дерматит и алопеция. Возможно развитие нарушений сознания, летаргии, комы на фоне метаболического ацидоза, лактатацидоза, иногда сочетающегося с гипераммониемией.
В случае поздней манифестации заболевания в клинической картине на первый план выступают атаксия, синдром мышечной дистонии в сочетании с дерматитом и алопецией [10]. Миелопатия может быть дополнительным симптомом заболевания, независимо от возраста его дебюта [21]. Нарушения функций органов чувств проявляются снижением слуха сенсоневрального генеза и зрения вследствие атрофии зрительных нервов. В единичных наблюдениях описаны дисплазия сетчатки, глаукома, миопия [13, 23]. Не исключено, что более позднему проявлению патологии способствует искусственное вскармливание ребенка в связи со значительно более высоким содержанием биотина в молочных смесях по сравнению с грудным молоком.
Недостаточность клеточного иммунитета приводит к различным осложнениям инфекционной природы: кандидозному кератоконъюнктивиту, диспепсическим нарушениям, частым респираторным заболеваниям.
Лабораторные и инструментальные критерии диагностики недостаточности биотинидазы. К основным методам диагностики недостаточности биотинидазы относятся биохимические и молекулярно-генетические методы. Основу развивающихся обменных нарушений составляет метаболический ацидоз, выраженность которого в значительной мере определяет тяжесть состояния пациентов. Ацидоз сопровождается кетозом, молочной и пировиноградной ацидемией, ацидурией. Повышена экскреция с мочой органических кислот: 3-гидроксиизовалериановой, 3-метилкротоновой, пропионовой, 3-гидроксипропионовой, 3-окси-2-метилмасляной, 3-метил-кротонилглицина.
Сноска 4. Для диагностики заболевания необходимо выявление специфической органической ацидурии и низкой активности биотинидазы в сыворотке крови.
Для диагностики заболевания необходимо выявление специфической органической ацидурии и низкой активности биотинидазы в сыворотке крови. В норме активность биотинидазы в сыворотке крови составляет 4,4-12 нмоль/мин/мл [6].
При электроэнцефалографическом (ЭЭГ) исследовании и компьютерной томографии (КТ) на ранних стадиях болезни нарушений может быть не выявлено. В более поздние сроки на ЭЭГ определяются двусторонние биоэлектрические изменения с характерными пиками высокоамплитудной активности. Для картины КТ характерны расширение желудочковой системы и борозд головного мозга, диффузные изменения белого вещества с признаками церебральной и церебеллярной атрофии, иногда нарушения в области хвостатого и чечевицеобразного ядра по типу геморрагического инфаркта. При МРТ головного мозга подтверждаются нарушения миелинизации, отек белого вещества, атрофические изменения коры.
Принципы лечения. Основным методом патогенетического лечения является пероральный прием биотина в дозе 6-40 мг в сутки. Дозу увеличивают постепенно. Препарат применяют в течение длительного времени. Терапию биотином можно начинать, не дожидаясь результатов определения активности биотинидазы в сыворотке крови. Единственным противопоказанием к терапии биотином является индивидуальная непереносимость. При наличии судорог применяют антиконвульсанты, но без одновременного применения биотина они оказываются недостаточно эффективными. Кроме того, следует учитывать, что на фоне приема антиконвульсантов снижается абсорбция биотина. Тем не менее практически всем пациентам с дефицитом биотинидазы назначаются антиконвульсанты из группы вальпроатов, принимая во внимание высокую частоту судорожных приступов и нередко их серийный и полиморфный характер. У детей раннего возраста применяют препарат конвулекс из группы вальпроатов: капли для приема внутрь (300 мг вальпроата натрия/мл, что соответствует 10 мг препарата в 1 капле, во флаконах по 100 мл) или сироп (50 мг/мл во флаконах по 100 мл).
Сноска 5. Основным методом патогенетического лечения является пероральный прием биотина в дозе 6-40 мг в сутки.
Сноска 6. Раннее начало лечения биотином в неонатальный период дает возможность предотвратить формирование тяжелых клинических проявлений и умственной отсталости.
Раннее начало лечения биотином в неонатальный период дает возможность предотвратить формирование тяжелых клинических проявлений и умственной отсталости [1,8,19,23].Позднее начало терапии приводит к исчезновению некоторых симптомов, таких как дерматит, алопеция, но не позволяет полностью купировать неврологические нарушения [20].
Приведем пример собственного наблюдения клинического случая неонатальной формы карбоксилазной недостаточности (дефицита биотинидазы).
Пациент К.А., 2,5 мес., в экстренном порядке доставлен в отделение психоневрологии в связи с повторными судорожными приступами, утратой психомоторных навыков, атопическим дерматитом, персистирующим конъюнктивитом со слизисто-гнойным отделяемым из глаз.
Из анамнеза: начало заболевания в возрасте 2 мес., когда на фоне полного здоровья во время бодрствования впервые возникли генерализованные миоклонические приступы, захватывающие все основные группы мышц, продолжительностью до 1 мин, с частотой 1 раз в сутки; отмечалось постприступное возбуждение. В течение недели наблюдалось учащение приступов до 2-3 раз в сутки, присоединение сложных парциальных приступов (с поворотом головы влево/вправо, адверсией глаз), продолжительностью до 1 мин, с частотой 1-2 раза в сутки. В связи с учащением генерализованных миоклонических и появлением сложных парциальных приступов до 6-8 раз в сутки пациент был госпитализирован в отделение психоневрологии, где ему была впервые назначена антиэпилептическая терапия (конвулекс до 75 мг/кг/сут) с незначительным положительным эффектом (урежение приступов на 25%).
На второй неделе госпитализации по данным физикального обследования, рентгенографии грудной клетки была диагностирована острая правосторонняя нижнедолевая пневмония с сопутствующим бронхитом. Ребенок переведен в инфекционное отделение. На фоне проводимой антиэпилептической терапии, инфузионной и антибактериальной терапии сохранялись сложные парциальные приступы с частотой 1-2 раза в сутки, присоединились серийные приступы по типу инфантильных флексорных спазмов (до 8 серий в сутки, по 5 приступов в серии).
Из анамнеза жизни известно, что мальчик родился от матери 29 лет c неотягощенным соматическим и акушерско-гинекологическим анамнезом, от I беременности, протекавшей физиологически, от I срочных родов на 39 нед., путем экстренной операции кесарево сечение (слабость родовой деятельности, стимуляция окситоцином; безводный период более 9 ч). Оценка по шкале Апгар 7/8 баллов. Кефалогематома в левой теменно-затылочной области 2,0 x 1,5 см. Вес при рождении 3520 г, рост 51 см. К груди приложен на 1 сутки. Неврологических нарушений в период пребывания в родильном доме не было. Выписан домой на 6 сутки. Лечебных рекомендаций не было. Психомоторное развитие до дебюта заболевания: держит голову в положении на животе с 1,5 мес., в вертикальном положении – с 2 мес. Следит за игрушкой, улыбается, гулит с 2 мес.
Соматический статус при поступлении: состояние тяжелое по основному заболеванию. Сопор. Ребенок отказывается от еды. Телосложение правильное. Кожные покровы бледные, сухие, крупнопластинчатое шелушение по всему телу. Тотальная алопеция. Периферические лимфоузлы не увеличены. Видимые слизистые оболочки обычной окраски, чистые. Конъюнктива глаз отечна, гиперемирована. Катаральных явлений нет. Дыхание стридорозное. В легких дыхание жесткое, проводится по всем полям, хрипов нет. Границы сердца в пределах нормы. Тоны сердца громкие, ритмичные. Живот мягкий, безболезненный. Печень и селезенка не увеличены. Стул неустойчивый, диурез в норме. Ребенок находится на естественном вскармливании.
Неврологический статус при поступлении: общемозговых и менингеальных симптомов нет. Реакция на осмотр вялая. Голову удерживает кратковременно. Коммуникативные навыки снижены. Не улыбается, не гулит. Сохранен ритм сна и бодрствования. Окружность головы 39,5 см. Форма черепа правильная. Большой родничок 2 см; малый родничок закрыт. Со стороны черепных нервов: обоняние ориентировочно сохранено. Взгляд фиксирует недолго, за предметами прослеживает непостоянно, кратковременно. Глазные щели симметричны, объем движений глазных яблок полный. Зрачки округлой формы, симметричные. Фотореакции живые. Трофика жевательной мускулатуры не нарушена. Лицо симметрично в покое, при плаче. Голос звонкий. Глотание не нарушено. Язык в полости рта по средней линии. Двигательно-рефлекторная сфера: спонтанная двигательная активность незначительно снижена. Диффузная мышечная гипотония. Мышечная сила в норме. Сухожильные рефлексы оживлены, симметричны. Брюшные рефлексы средней живости, симметричные. Рефлекс Бабинского положителен с двух сторон. Сохранены рефлексы периода новорожденности: асимметричный шейный тонический рефлекс, Моро, Робинзона. Вегетативная нервная система:дермографизм – красный, стойкий, гипергидроза нет. Функции тазовых органов не нарушены.
Данные лабораторных и инструментальных методов обследования при поступлении:
· в клинических анализах крови и мочи в динамике отклонений от нормы не выявлено;
· показатели кислотно-основного состояния крови: при поступлении выраженный метаболический ацидоз: рН капиллярной крови 7,22 (норма 7,37-7,45), АВЕ -15,6 ммоль/л (норма 0±2,3), pCO2 27,6 мм рт. ст. (норма 32-45), pO2 71,9 мм рт. ст. (норма 83-108), HCO3 - 10,9 ммоль/л (норма 22,2-27,9);
· кровь на содержание лактата: концентрация лактата повышена - 4,8 (норма 0,9-1,8);
· кровь на аминоацидурии: выявлено повышение концентрации 3-гидрокси-изовалерил-2-метил-3-гидроксибутирилкарнитина (недостаточность биотинидазы);
· кровь на определение активности биотинидазы: активность 0,26 нмоль/мин/мл (норма 4,40-12);
· исследование внутренних органов: выявлены диффузные изменения поджелудочной железы. Печень и почки – без патологии;
· ЭКГ: нерезкая синусовая тахикардия (при беспокойстве ребенка). Вертикальная электрическая ось сердца;
· компьютерная томография головного мозга: срединные структуры не смещены. Желудочки не деформированы. Конвекситальные борозды умеренно расширены. Латеральные щели широкие, симметричные. Межполушарная щель расширена до 0,6 см. Выраженное снижение плотности вещества головного мозга перивентрикулярно и в лобных отделах;
· осмотр окулиста: ОU - слизисто-гнойное отделяемое из глаз. Конъюнктива гиперемирована. Глазные щели симметричны. Девиации нет. Подвижность глаз не ограничена. Оптические среды прозрачны. Глазное дно: диски зрительных нервов светло-розовые, контуры четкие. Ход и калибр сосудов не изменены. Макулярная зона и периферия без патологии. Заключение: острый бактериальный конъюнктивит.
Лечение: в течение 2 недель без значительного эффекта проводилась терапия дексазоном, витамином В6, конвулексом до 75 мг/кг/сут, элькаром, к которой был дополнительно назначен биотин 10 мг/сут, с последующим увеличением дозы до 30 мг/сут. На вторые сутки применения патогенетической терапии биотином судороги были полностью купированы. После этого было начато постепенное снижение дозы конвулекса. Ребенок стал осваивать соответствующие возрасту моторные навыки, улучшились показатели его психического развития.
При динамическом наблюдении через 6 мес. в возрасте 8,5 мес. на фоне проводимой патогенетической терапии отмечалась положительная динамика: судороги не возобновлялись, ребенок активный, уверенно держит голову, следит за игрушкой, улыбается, тянется к игрушке, перекладывает из руки в руку, поворачивается со спины на живот и с живота на спину в обе стороны, среди окружающих людей узнает своих и чужих, гулит, встает на четвереньки. Получает терапию: биотин 30 мг/сут, конвулекс 15 мг/кг/сут, элькар.
Действующим веществом препарата элькар является L-карнитин (левокарнитин), которое представляет собой природное биоактивное витаминоподобное вещество, играющее важнейшую роль во всех обменных процессах, активно участвующее в образовании энергии и являющееся универсальным анаболическим средством негормональной природы. Элькар является препаратом системного действия и оказывает положительное влияние на основные звенья патогенеза нарушений нервно-психического развития детей различного возраста, в т.ч. раннего. Кроме того, элькар рекомендуется включать в комплексное лечение судорожных состояний у детей, особенно при использовании вальпроатов.
Массовый скрининг новорожденных появился в развитых странах мира с конца 1960-х гг., когда стало проводиться обследование на фенилкетонурию. В рамках различных программ во многих странах стало проводиться тестирование и на другие наследственные болезни. Однако новая эра в развитии неонатального скрининга была открыта с появлением тандемной масс-спектрометрии [9] – технологии, существенно повышающей качество диагностики и позволяющей значительно расширить список выявляемых заболеваний, которые поддаются лечению, но не во всех случаях обнаруживались с помощью методов, ранее применявшихся в программах скрининга.
Читайте также: