
Синдром канавана кратко и понятно
Обновлено: 30.06.2024
Проявляющееся в раннем детстве неврологическое заболевание, сопровождающееся нарушением моторики, гипотонией мышц, приступами эпилепсии, слепотой и макроцефалией, характеризующееся дефицитом фермента аспартоацилазы, называется спонгиозной дегенерацией, или болезнью Канавана.
По какой причине развивается болезнь Канавана?
Развитие патологии ведёт к демиелинизации нервных волокон головного мозга. Заболевание имеет генномутационную природу развития. Необходимо отметить, что данная патология относится к разряду редко встречаемых неврологических заболеваний. В подавляющем большинстве случаев заболевание развивается на первом году жизни ребёнка. К сожалению, приходится констатировать неутешительные результаты современных методов терапии. Неврологи относят патологию к разряду лейкодистрофии. Причиной развития патологии является мутация гена ASPA, который находится на семнадцатой хромосоме, отвечающего за выработку фермента аспартоацилазы, расщепляющего отдельные молекулы аспарагиновый кислоты, формирующейся в тканях нервной системы. Дефицит фермента неизбежно ведёт к переизбытку аспарагиновой кислоты, в следствие чего происходит разрушение миелиновой оболочки. Данный патологический процесс оставляет нервные клетки незащищенными и значительно ухудшает передачу нейронных импульсов. Болезнь Канавана относят к группе дегенеративных неврологических заболеваний.
В качестве основных симптомов болезни Канавана выделяют:
- нарушения в фиксации взгляда;
- повышенную возбудимость;
- нарушение глотательной функции;
- приступы судорог;
- пониженный тонус мышц;
- нарушение моторики;
- запоздалое развитие ряда навыков;
- макроцефалию;
- патологии стоп и кистей.
Как правило, к двухлетнему возрасту атрофируется зрительный нерв, что приводит к полной потере зрения.
Высокую концентрацию аспарагиновой кислоты позволяет установить общий анализ мочи. Для точной диагностики необходимо провести биохимический анализ крови, диагностику ДНК, магнитно-резонансную томографию. После подтверждения диагноза болезни Канавана невролог назначает симптоматическую терапию.
Как мы уже говорили выше, прогноз неутешительный. В большинстве случаев летальный исход наступает до достижения ребёнком десятилетнего возраста. Сегодня в специализированных клиниках проводят искусственную замену матированного гена на здоровый. Это позволяет значительно продлить жизнь больному, однако летальный исход неизбежен. Все, чего пока удалось добиться врачам в области лечения болезни Канавана — это увеличить продолжительность жизни больного до двадцати лет. На сегодняшний день отсутствует также специфическая профилактика данной патологии.

Заболевания
Болезнь Канавана
Белое вещество – это ткань головного мозга которая в основном состоит из миелинизированных аксонов, которые представляют собой длинные ретрансляторы, выходящие из сомы, и которые имеют белый цвет из-за относительно высокого содержания липидного жира в миелиновом белке, который их покрывает. Миелин имеется и в центральной, и в периферической нервных системах. В ЦНС он находится преимущественно в белом веществе (хотя некоторые его количества имеются и в сером), как раз придавая ему такой цвет. Миелин, который можно назвать миелиновой оболочкой, защищает нервные волокна, действует как изолятор и увеличивает скорость передачи нервных сигналов. Каждый тип лейкодистрофии поражает разные части миелиновой оболочки, вызывая ряд различных неврологических проблем.
История
Эпидемиология
Симптомы
Дополнительные симптомы, которые поражают детей с болезнью Канавана, включают судороги, нарушения сна, трудности с кормлением, контрактуры крупных суставов, носовую регургитацию, гастроэзофагальный рефлюкс, и атрофию зрительных нервов что приводит к слепоте. В большинстве случаев это не влияет на слух, но могут и поражаться нервные волокна отвественные за слух. В последние несколько лет была выявлена легкая форма болезни Канавана с типичными мутациями в гене ASPA и при этом незначительным увеличением NAA в моче. Эти дети имеют негрубую задержку психоречевого развития, однако они могут учиться и ходить в школу. Голова может быть несколько увеличена, но типичные изменения белого вещества, связанные с болезнью Канавана, могут отсутствовать. Прогноз, безусловно, намного лучше, чем при класической форме.
Генетические особенности
Это аутосомно-рецессивное заболевание, вызванное мутацией гена на коротком плече 17й хромосомы. Ген ASPA синетзирует фермент аспартоацилаза. Этот фермент расщепляет N-ацетиласпартат (NAA), который находится в нейронах головного мозга. Функция NAA неясна. Изначально предпологали что он играет роль в производстве миелиновой оболочки, но недавние исследования показывают, что NAA не имеет этой функции. Вместо этого фермент может участвовать в транспортировке молекул воды из нейронов. Мутации в гене ASPA снижают функцию аспартоацилазы, которая предотвращает нормальный распад NAA. Если NAA не расщепляется должным образом, возникающий в результате химический дисбаланс мешает формированию миелиновой оболочки по мере развития нервной системы. Накопление NAA также приводит к прогрессивному разрушению существующих миелиновых оболочек, что нарушает нормальное развитие мозга.
Дифференциальная диагностика
- Болезнь Александера;
- Болезнь Пелицея-мерцбахера;
- Адренолейкодистрофия;
- Метахроматическая лейкодистрофия;
Терапия
Клинический случай
В начале 2021 года к нам к клинику поступил мальчик в возрасте 4 месяцев, с основными жалобами при поступлении: ежедневные серийные судорожные приступы, грубая задержка психомоторного развития, отсутствие пищевого поведения, комплекса оживления, снижение массы тела.
Анамнез заболевания: ребенок от 2 беременности (сестра 2,5 года – здорова), с ОРВИ в 20 недель, носительство коронавирусной инфекции в 38 нед. Роды путем планового кесарева сечения в 38 недель. При рождении масса 2770гр, длина 52 см, Апгар 8/9 баллов. Выписан из роддома на 17 с/ж в связи с перинатальным контактом по COVID. По данным НСГ определялись постгипоксичесике (?) Изменения. В 1 месяц осмотрен неврологом – перинатальное поражение ЦНС с нервно-рефлекторной возбудимостью.
В 2 месяца – окружность головы +10 см от рождения, однако по НСГ признаков повышения внутричерепного давления не отмечалось.
Проведено КТ головного мозга по месту жительства – признаки перивентрикулярной лейкомаляции.
Так же по месту жительства проведено МРТ головного мозга где были выявлены признаки лейкодистрофии.
Исключен синдром Ван дер Кнаап молекулярно-генетическим методом.
Данные объективного осмотра: Состояние пациента тяжелое. В сознании. Ребенок не контактен. Положение вынужденное. Вес/масса тела: 7 кг. (перцентиль 50-75%). Индекс массы тела: 16,57. Рост/длина: 65 см. (перцентиль 90-97%). Площадь поверхности тела: 0,36 кв.м. Окружность головы: 45 см. (перцентиль> 97%). Окружность груди: 44 см. (перцентиль 75-90%). Физическое развитие: очень высокое. Тип телосложения гармоничный. Имеются единичные микроаномалии. Кожа: сухая, на щеках – мелкоточечная сыпь. Слизистые оболочки не изменены. Подкожно-жировая клетчатка развита удовлетворительно. Лимфатические узлы множественные, мелкие, эластичные, безболезненные. Мышечная система развита удовлетворительно. Тонус мышц: нормальный. Форма грудной клетки правильная. Костные деформации не выявлены. Утолщение ногтевых фаланг пальцев нет. Суставы не изменены. Частота дыхания: 32 в мин. Одышки нет. Носовое дыхание свободное. Голос не изменен. Кашель не отмечается. Мокроты нет. Кровохарканье нет. Перкуторный звук не изменен. Дыхание: жесткое. Хрипы не слышны. Пульс: 120 в мининуту, ритм правильный. А/Д (прав.рука): 85/55 мм.рт.ст. Пульс на лучевых артериях нормальный. Перкуссия сердца границы соответствует возрасту верхняя – не изменена правая – не изменена левая – не изменена. Тоны сердца: отчетливые, ритмичные, выслушивается шум. Фаза сердечного цикла: систолическая. Аппетит: удовлетворительный, принимает противосудорожную тепатию. Тошноты нет. Рвоты нет. Другие диспептические явления не выявлены. Язык: чистый. Склеры: не изменены. Живот: мягкий, безболезненный. Симптомы желчного пузыря: отрицательные. Точки проекции поджелудочной железы: безболезненные. Асцита нет. Печень пальпируется. Край закруглен, эластичный. Селезенка: не пальпируется. Симптомы раздражения брюшины: нет. Стул не изменен. Мочеиспускание безболезненное. Дизурические явления не выявлены. Симптом поколачивания отрицательный. Вторичные половые признаки соответствуют возрасту. Осмотр половых органов сформированы правильно по мужскому типу. Психическое развитие: грубая задержка психо-речевого развития. Черепные нервы: Реакция зрачков на свет живая, зрачки симметричны. Глазные щели симметричны. За предметом не прослеживает, взгляд фиксирует кратковременно.
Сосание, глотание присутствуют, периодически поперхивается пищей. Корнеальный рефлекс вызывается. Лицо в покое и при плаче симметрично, мимика скудная. Мягкое небо симметрично, глоточный рефлекс живой. Язык в полости по средней линии, Кривошеи нет. Общая двигательная активность снижена.
Положение рук и ног свободное. Пронация и супинация кистей рук полная. Тонус мышц в руках в норме D=S. Тонус мышц в ногах в норме. D=S. Сухожильные рефлексы оживлены. При осмотре отмечаются серийные симметричные спазмы, миоклонии левых конечностей с заведением глаз с предшествующей икотой.
На момент госпитализации: Дебют эпилепсии – отведение глаз вправо, клонические подергивания в руках. На этом фоне была назначена противосудорожная терапия, с положительной динамикой в виде снижения числа приступов.
При видео ЭЭГ мониторинге выявлена диффузная эпилептиформная активность в виде одиночных комплексов острая-медленная волна амплитудой до 120 мкв.
По данным лабораторных исследований, Общего клинического анализа крови, исследования показателей основного обмена, биохимического исследования крови, значимых изменений показателей не выявлено.
Сердечно сосудистая система без патологических изменений.
Врач-офтальмолог – косоглазие расходящееся непостоянное.
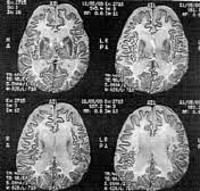
На МРТ головного мозга – Отмечается диффузное поражение белого вещества больших полушарий головного мозга. Имеет место ограничение диффузии в пораженном белом веществе. На этом фоне отмечается уменьшение объема серого вещества. Базальные ядра несколько увеличены, вблизи чечевицеобразных ядер с обеих сторон визуализируются небольших размеров ликворные кисты. Очаговых изменений в стволе и мозжечке не выявлено. Срединные структуры головного мозга не смещены. Мозолистое тело истончено на всем протяжении. Ликворная система не изменена.
Болезнь Канавана (спонгиозная младенческая дегенерация) — генетическое нейродегенеративное заболевание, в основе которого лежит недостаток фермента аспартоацилазы, приводящий к демиелинизации нервных волокон головного мозга. Болезнь Канавана проявляется в раннем детском возрасте нарушениями моторики и развития ребенка, затруднениями при приеме пищи, мышечной гипотонией, макроцефалией, эпилептическими припадками, слепотой. Диагностируется болезнь Канавана по анализу мочи на N-ацетил-аспарагиновую кислоту. Эффективная терапия заболевания пока не найдена. Производятся попытки лечить болезнь Канавана при помощи метаболической терапии и путем применения генных технологий, способных заменить патологическую аберрацию на здоровый ген.
МКБ-10
Общие сведения
Болезнь Канавана была описана Миртелем Канаваном в 1931 году. А в 1991 году Рубеном Маталоном был выделен ген, отвечающий за это заболевание. Спустя несколько лет стал применяться тест, позволяющий диагностировать болезнь Канавана еще в период ведения беременности. Болезнь Канавана встречается у людей любой национальности. Однако чаще всего ею болеют евреи ашкенази. Среди них носителем генной аберрации, кодирующей болезнь Канавана, является 1 из 40 человек.
Современная неврология относит болезнь Канавана к так называемым лейкодистрофиям — генетически обусловленным дегенеративным заболеваниям нервной системы, в основе которых лежит нарушение метаболизма липидов. Чаще всего нарушения касаются миелина, который на 70-75% состоит из липидов. Распад миелина приводит к разрушению миелиновой оболочки нервных стволов и вторичной гибели нейронов. Лежащая в основе заболевания прогрессирующая демиелинизация делает болезнь Канавана и другие лейкодистрофии схожими с демиелинизирующими заболеваниями головного мозга, которые имеют приобретенный характер (рассеянным склерозом, рассеянным энцефаломиелитом, полирадикулоневропатией Гийена-Барре, болезнью Девика).
Причины возникновения болезни Канавана
Болезнь Канавана связана с генетическим нарушением в геноме ASРА, расположенном в 17-й хромосоме. Вследствие этой генетической аберрации снижен синтез фермента аспартоацилазы, расщепляющего N-ацетил-аспарагиновую кислоту, образующуюся в процессе жизнедеятельности клеток центральной нервной системы. Патологически высокий уровень этой кислоты приводит к повреждению миелиновой оболочки нервных волокон головного мозга. Миелиновая оболочка выполняет роль своеобразного изолятора, не позволяющего нервному импульсу переходить с одного нервного волокна на другое. Таким образом, миелиновая оболочка способствует точному и быстрому проведению нервных импульсов. При ее разрушении этот процесс нарушается, что и является непосредственной причиной клинических проявлений болезни Канавана.
Болезнь Канавана передается по аутосомно-рецессивному типу наследования. Это означает, что в ситуации, когда оба родителя являются носителями патологического гена, вероятность рождения у них ребенка, имеющего болезнь Канавана, равна 25%.
Симптомы болезни Канавана
При рождении ребенок, имеющий болезнь Канавана, ничем не отличается от здоровых новорожденных. Симптомы заболевания начинают проявляться спустя месяц, а чаще в период от 3 месяцев до полугода. Отмечается задержка в развитии, замедленность движений, потеря уже приобретенных двигательных навыков, снижение мышечного тонуса. Возникающие затруднения при глотании обуславливают существенные трудности при кормлении ребенка. Примерно к 4-ому месяцу жизни становиться заметно увеличение головы (макроцефалия). Прогрессирование болезни Канавана сопровождается возникновением эпилептических припадков. Демиелинизация и атрофия зрительного нерва приводит к развитию слепоты.
Диагностика болезни Канавана
Болезнь Канавана представляет для невролога определенные сложности в диагностике, поскольку демиелинизирующий процесс наблюдается при целом ряде как врожденных, так и приобретенных заболеваний. Стандартные методы первичного неврологического обследования, такие как электроэнцефалография и ультрасонография не выявляют специфических изменений. Проведение МРТ головного мозга затруднено из-за слишком раннего возраста пациентов. Наиболее достоверным методом, позволяющим диагностировать болезнь Канавана, является анализ мочи на N-ацетил-аспарагиновую кислоту.
Генетики рекомендуют всем лицам еврейской национальности при планировании беременности проходить скрининг на наличие генной мутации, определяющей болезнь Канавана. Скрининг следует проходить также тем людям, в семье которых наблюдался случай этого заболевания. Разработаны методы пренатальной диагностики, позволяющие установить болезнь Канавана еще до рождения ребенка. Показанием к проведению подобного исследования является установленное носительство патологического гена у обоих родителей. Материалом для исследования на болезнь Канавана служат амниотическая жидкость, полученная в результате амниоцентеза, или ворсины хориона, взятые в ходе трансабдоминальной или трансцервикальной биопсии хориона.
Лечение болезни Канавана
В настоящее время не существует методики, при помощи которой можно было бы эффективно лечить болезнь Канавана. Замедлить развитие заболевания можно при применении метаболической терапии. Она заключается в приеме внутрь целого комплекса лекарственных препаратов: цитрата лития, ацетата кальция, сукцината натрия.
Наиболее оптимистичными среди современных экспериментальных разработок в области лечения болезни Канавана является генная терапия. Разработка метода генной терапии, способного значительно продлить жизнь имеющим болезнь Канавана пациентам, проводится в Нью Джерси (США). Ее идея заключается в клонировании и внедрении в организм здорового гена, способного заменить патологический ген. Носителями нового искусственно синтезированного гена являются непатогенные аденоассоциированные вирусы, которые в растворе водятся в головной мозг через 6 катетеров, установленных в различных его областях. Экспериментальное лечение прошли 13 детей. Их последующее обследование показало, что в результате генной терапии болезнь Канавана замедлила свое прогрессирование, произошло снижение токсичной N-ацетил-аспарагиновой кислоты и увеличение содержание миелина.
Прогноз болезни Канавана
К сожалению, болезнь Канавана приводит к летальному исходу еще в младенческом возрасте. В редких случаях больные доживают до возраста 10 лет.
Болезнь Канавана — это генетическая аутосомно-рецессивная нейродегенеративная болезнь, известная также под названиями Болезнь Канавана-Ван Богерта-Бертрана. Это спонгиозная дегенерация белого вещества мозга, спонгиозная младенческая дегенерация, при которой наблюдается прогрессирующее поражение нервных клеток головного мозга. Аутосомно-рецессивный тип наследственности значит, что ребенок, родившийся от родителей-носителей данного заболевания, имеет высокий риск заболеть (примерно 25%). Это заболевание редкое, но в то же время одно из наиболее распространённых у детей, связанное с нарушением работы мозга. Принадлежит к группе генетических заболеваний под названием лейкодистрофия.
Проявляется болезнь в раннем детском возрасте, распространена в большей степени среди еврейского населения ашкенази.
Причины возникновения болезни Канавана
Болезнь Канавана вызывается мутировавшим геном ASРА, который синтезирует фермент аспартоацилазы и находится в семнадцатой хромосоме.
В головном мозге в высокой концентрации из аспаргиновой кислоты образуется ацетиласпарагиновая кислота и присутствует там так же, как и глутаминовая кислота.
Роль и значение этой кислоты до конца не определено, но известно, что недостаток аспартоациллазы приводит к возникновению болезни Канавана. Тогда эта кислота выводится из организма с мочой, повышая ее концентрацию в разы. Большое количество этой кислоты появляется в крови и самом головном мозге, особенно в белом веществе, где наблюдается резкое набухание астроцитов.
При электронной диагностике выявляется деформация митохондрии. В головном мозге происходят прогрессирующие атрофические процессы, в следствие которых увеличиваются в объеме мозговые желудочки. Нарушается образование миелиновой оболочки (особенный слой жиров, покрывающий нервные клетки как бы изолируя их, не позволяя нервному импульсу переходить из одного нервного волокна в другое).
Симптомы болезни Канавана
Болезнь Канавана изначально может никак не проявляться. Только спустя месяц или три месяца после рождения ребенка вероятно поставить окончательный диагноз.
К симптомам болезни Канавана относятся:
- развитие мышечной гипотонии (ребенок не держит голову);
- повышенная сонливость;
- снижение мышечного тонуса шеи и увеличение тонуса мышц конечностей;
- тугоподвижность суставов;
- наличие судорог;
- хаотическое движение глазных яблок;
- эпилептические припадки;
- пищевой рефлюкс, нарушение функции глотания;
- паралич конечностей;
- увеличение головного мозга и образование в нем полостей;
- атрофия зрительных нервов — слепота (к двухлетнему возрасту);
- макроцефалия — увеличение головы вследствие накопления жидкости;
- тяжелое отставание в развитии.
Симптомы болезни Канавана быстро прогрессируют, поэтому продолжительность жизни пациентов сокращается. Конечная стадия развития заболевания характеризуется обездвиживанием пациента, затруднительным дыханием и кровообращением.
Диагностика болезни Канавана
Диагностика болезни Канавана — сложный процесс, потому что похожие симптомы наблюдаются и среди других врожденных или приобретенных болезней.
Для определения болезни Канавана или подтверждения диагноза проводится тщательная диагностика, включающая в себя:
- анализ крови и органический анализ мочи на кислотность;
- КТ;
- МРТ;
- генетический тест на генные мутации.
Анализ мочи и крови сразу выявляет высокую концентрацию ацетиласпарагиновой кислоты. Процесс проведения МРТ затруднителен, так как возраст пациентов очень мал, поэтому могут возникать неточности в диагностике. МРТ помогает выявить губчатую дегенерацию нервных волокон миелинового жирового слоя, а также набухание астроцитов и деформацию (удлинение) митохондрий. Концентрация аспартоацилазы в кожных фибробластах у носителей болезни Канавана приблизительно в два раза ниже нормы. Магнитно-резонансная спектроскопия показывает наивысшую концентрацию ацетиласпарагиновой кислоты в головном мозге. Особенно важен биохимический анализ крови и мочи, которые и дают заключение диагноза, играют решающую роль.
Существуют методы диагностики, позволяющие выявить данное заболевание ребенка еще в утробе матери (перинатальная диагностика). Для этого нужно установить в первую очередь деформированный патологический ген у обоих родителей. Для подтверждения или опровержения диагноза проводится амниоцентез для получения амниотической жидкости. Практикуют также трансабдоминальную или трансцервикальную биопсию хориона для получения ворсины хориона.
Генетики рекомендуют лицам, имеющим в семейной истории данное заболевание, при планировании семьи проходить тщательное обследование — скрининг на выявление модифицированного гена.
Лечение болезни Канавана
Эффективной и стандартной методики, как и лекарств для лечения болезни Канавана, на сегодняшний день не существует. Курс лечения сугубо индивидуален и имеет поддерживающий характер. Болезнь считается смертельной. Пациенты умирают зачастую в возрасте до двух лет, в отдельных случаях продолжительность жизни может быть до десяти лет, в редких случаях до двадцати. С помощью метаболической терапии возможно замедление развития болезни. Прием ряда медицинских препаратов (ацетат кальция, цитрат лития, сукцинат натрия, милидиамокс) сможет приостановить прогрессирование заболевания, но остановить его не сможет. Все препараты, необходимые для лечения — дорогостоящие.
Экспериментальное лечение
Эксперименты медиальной терапии на основе нитрата лития (проводились в лабораторных условиях на организмах животных) дали обнадёживающие результаты приостановления заболевания. Это стало основой для продолжения экспериментального лечения на основе лития.
На уровне экспериментов исследуются разнообразные методы лечения, в частности, генная терапия. Например, в желудочки мозга пациента через несколько катетеров были введены липосомы с геном аспартоациалазы в качестве раствора. Процедура проводилась как экспериментальное лечение с участием более десятка больных детей и не привела к желаемому результату — полное устранение болезни, хотя и считается наиболее успешной, так как с ее помощью удалось приостановить развитие заболевания.
Обнадёживающие результаты дали эксперименты с использованием триацетина, который расщепляют до ацетата для лучшего усвоения организмом.
Прогноз болезни Канавана

Вряд ли нужно объяснять, какая это беда не только для ребенка, но и для его родителей. Поэтому очень важно знать, из-за чего возникает такое заболевание, можно ли его предотвратить и что следует делать, если оно, к несчастью, диагностировано.
В чем причина возникновения болезни Канавана и как она диагностируется
Другие методы исследования, вплоть до МРТ и электроэнцефалографии, затруднительно производить, и к тому же они не дают должной эффективности ввиду малого возраста пациента.
Общие сведения об этом заболевании
Читайте также: