
Кислотно основной катализ кратко
Обновлено: 03.07.2024
Кислотно-основной катализ (кислотно-основный катализ), ускорение хим. реакций в присутствии кислот и оснований. В качестве катализаторов используют: в гомог. кислотном катализе протонные кислоты (H 2 SO 4 , HCl, Н 3 РО 4 , CH 3 C 6 H 4 SO 2 OH и др.) в воде и водно-орг. растворителях, апротонные кислоты (АlС1 3 , BF 3 , SnCl 4 и др.) в неводных растворителях, сверхкислоты (HF SbF 5 , HSO 3 F SbF 5 и др.) в неводных растворителях; в гетерог. кислотном катализе прир. глины. аморфные и кристаллич. алюмосиликаты. фосфорную и полифосфорные кислоты, нанесенные на носитель, катиониты. в гомог. основном катализе оксиды щелочных металлов. амины в воде, орг. и водно-орг. растворителях; в гетерог основном катализе - оксиды металлов (CaO, MgO и др.). При К.-о. к. в большинстве случаев из реагентов и катализатора в равновесных стадиях образуются реакционноспособные (а также нереакционноспособные) комплексы разл. состава, которые являются ионизир. формой реагента. В лимитирующих стадиях комплексы превращ. в продукты реакции. В случае реакции с одним реагентом (Р) эти стадии выражаются ур-нием:
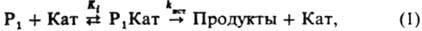
где Кат-разл. формы катализатора: недиссоциир. кислоты или основания, ионы или ионные пары. образующиеся при их ионизации; P 1 Кат-комплексы; К i - константа равновесия образования комплексов Р 1 Кат; k ист - элементарная константа скорости превращ. комплексов в продукты. В случае реакции двух реагентов (P 1 + Р 2 : Продукты; Р 2 +Р 2 : Продукты) реакционноспособный комплекс образуется, как правило, из комплекса одного реагента с несвязанным в комплекс др. реагентом:
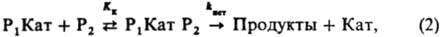
где К к - константа равновесия образования комплексов Р 1 Kат Р 2 . Константы равновесия K i и К к выражают ур-ниями:

где (здесь и далее) а, с, f -соотв. термодинамич. активность, концентрация и коэф. активности реагирующих частиц; a Kат f P1 /f P1Кат - ф-ция, характеризующая способность среды переводить реагенты в комплексы РКат. При варьировании в широких пределах с Кат реализуются (особенно в растворах) неск. маршрутов реакции - хим. превращений, приводящих к образованию одних и тех же продуктов через реакционноспособные комплексы (РК) разл. состава. В растворе с постоянной с Кат скорость реакции по каждому маршруту W i выражается ур-нием:
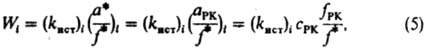
где (k ист ) i - элементарная константа скорости по i-му маршруту, а* и f* - относятся к активир. комплексу. Из опытных данных следует, что множитель f РК /f* не изменяется при варьировании с Кат и его приравнивают к единице. В растворе с постоянной с Кат наблюдаемая скорость реакции
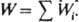
Экспериментально определяют эффективную константу скорости k эфф , характеризующую скорость реакции при постоянной температуре и наличии катализатора определенного состава. В ур-ниях для связи f эфф со свойствами катализатора учитывают все его комплексы с реагентом: реакционноспособные (РКат) РК и нереакционноспособные (РКат) НРК . Напр., при кислотном гидролизе амидов, анилидов ионизированная по атому азота форма является реакционноспособной, ионизированная по карбонильной группе - либо намного менее реакционноспособна, либо нереакционноспособна. Для описания влияния ионизирующих свойств среды на k эфф совместно решают ур-ние материального баланса для текущих концентраций реагентов и ур-ния (3) и (5) в случае реакции одного реагента и дополнительно ур-ние (4) для реакции двух реагентов. например для реакции, протекающей по схеме (2):

В случае реакции двух реагентов, если один (Р 2 ) является компонентом растворителя (напр., Н 2 О при гидролизе в водных растворах), при условии с р2 >>е р1 вычисляют эффективную константу скорости первого порядка, включающую термодинамич. активность растворителя. В реакциях Р х с Р 2 в растворах кислот и оснований реакционноспособный комплекс состава P 1 KaтP 2 может получаться и в лимитирующей стадии из ионизир. формы одного реагента и неионизир. формы другого:
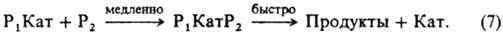
В этом случае для описания влияния ионизирующих свойств среды на k эфф пригодно ур-ние (6), при замене величины (f P2 с P2 )/(К к f P1КатP2 ) на термодинамич. активность растворителя. Поскольку крайне редко удается установить прямыми физ. методами состав и концентрацию промежут. комплексов, в настоящее время основным методом установления их состава, детального механизма реакции и вычисления величин К i и К к является кинетич. метод, основанный на применении ур-ний типа (6) для объяснения наблюдаемой зависимости k эфф от ионизирующей способности среды. Для этого измеряют k эфф в широком диапазоне с Кат в растворе и подбирают ф-ции а Кат f P /f PКат для описания влияния среды на k эфф . наиб. исследованы каталитич. свойства сильных кислот в водных растворах. Ионизирующая способность таких растворов обусловлена образованием комплексов реагента: 1) с сольватированным протоном и 2) с недиссоциированной кислотой. К первым относятся протонир. форма реагента и ее комплексы с растворителем. В этом случае а Кат f P /f PKат выражают (для водных растворов) ф-циями h 0 =a H3O+ f P /f PH+ , h 0 a H2O и h 0 a 2 Н2O , где h 0 - измеренная индикаторным методом кислотность среды, f РН+ -коэф. активности протонир. формы реагента. Ко вторым относятся комплексы реагента с кислотой (НА) и ее гидратами. При этом a Кат f Р /f РКат кат принимает значения а НА , a НА a Н2O и др. При гидролизе некоторых карбонильных соед. a Кат f P /f РКат =c H5O2 + Однозначную информацию о составе реакционноспособных комплексов, как правило, получают на основании данных по изменению k эфф в растворителе постоянного состава при варьировании с Кат в широких пределах в условиях малой степени связывания реагента в комплексы. наиб. простые зависимости получают для реакции одного реагента, когда он равновесно связывается только в один реакционноспособный комплекс. В таком случае ур-ние (4) упрощается:

Опытным путем установлено, что в водных растворах сильных кислот k ист не зависит от с Кат . К i , по определению, не должно зависеть от с Кат . Вопрос о влиянии состава смешанного растворителя на k ист пока достаточно не исследован. В условиях, когда K i и (a Кат f Р )/f РКат соизмеримы, по ур-нию (8) вычисляют раздельно k ист и K i . При а Кат f Р /f РКат >> K i величина k эфф =k ист ; при k Кат f P /f РКат i имеет место соотношение:
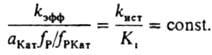
Ниже приведен пример, раскрывающий природу каталитич. действия водных растворов кислот в условиях, когда реакционноспособный комплекс образуется и превращ. в продукты согласно схеме (1) при условии, что а Кат f P /f РКат i . Ионизирующая способность среды из-за превращ. реагента в протонир. форму и ее комплексы с Н 2 О проявляется при наличии зависимостей К эфф /h 0 = const, k эфф (h 0 a H2О )=const k эфф /(h 0 a 2 H2O )=const. Если в реакции k эфф /h 0 =const (lgk эфф +H 0 =const, где H 0 =-lgh 0 - ф-ция кислотности Гаммета), то a кат f P /f Кат =h 0 и реакционноспособным комплексом является протонир. форма реагента РН 3 О + . Такая закономерность имеет место при проведении пинаколиновой перегруппировки в конц. H 2 SO 4 :

При варьировании с Kaт значение k эфф /(h 0 а H2O ) постоянно в реакциях дегидратации, например 2-фенил-2-пропанола в серной кислоте, 2-метил-2-бутанола в азотной кислоте. Следовательно, а Kат f P /f PKaт равно h 0 а H2O и реакционноспособным является комплекс молекулы воды с протонир. формой реагента. Имеется и пример постоянства k эфф /(h 0 a 2 H2O ), что наблюдается в реакции изотопного обмена атома кислорода в СН 3 ОН, протекающего в серной и хлорной кислотах. В этом случае а Kaт /f P /f РКaт =h 0 a 2 H2O и реакционноспособным является симметричный комплекс:
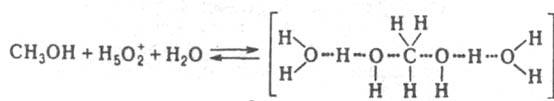
Ф-ции h 0 , h 0 а H2O и h 0 а 2 H2O стандартизованы к воде, и в сильно разбавленных растворах сильных кислот они численно равны концентрации ионов Н 5 О + 2 в моль/л. В таких растворах для приведенных выше реакций при варьировании с Kaт должно соблюдаться постоянство значений k эфф /c H2O + . При гидролизе некоторых амидов, анилидов, сложных эфиров k эфф /c H2O + постоянно не только в разбавленных, но и в конц. водный растворах НСlO 4 , H 2 SO 4 . Если гидролиз протекает по схеме (1), то реакционноспособный комплекс образуется из реагента и иона Н 5 О 2 + , а выраженная в концентрациях константа равновесия K H5O2 + ; не изменяется при варьировании с Кат :
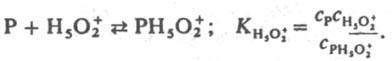
Не исключено, что k эфф / c H2O + =const из-за того, что реакция протекает по схеме (7) Ионизирующая способность среды из-за комплексообразования реагента с молекулами недиссоциир. сильной кислоты и ее гидратом проявляется при наличии зависимостей k эфф /a HA =const, k эфф /(a HA a H2O )=const и др. В первом случае a Kaт = const и реакци онноспособный комплекс имеет состав РИА; такая закономерность, например, реализуется при дегидратации b-фенил-b-гидроксипропионовой кислоты и 2-(4-мето-ксифенил)-2-пропанола в серной кислоте. Во втором случае а кат f P /f PKат =а HА а H2O и комплекс имеет состав РНАН 2 О. Гидрат НА.Н 2 О можно рассматривать и как ионную пару Н 3 О + А - . Каталитич. активность гидратов сильных кислот проявляется в реакциях дегидратации 1-фенилэтанола в хлорной кислоте, гидратации цис -коричной кислоты и рацемизации b-фенил-b-гидроксипропионовой кислоты в серной кислоте. Согласно ур-нию (6), должен наблюдаться максимум k эфф в зависимости от с Кат , если наряду с реакционноспособными образуются и нереакционноспособные комплексы и при увеличении с Кат значение a Кат /f P /f PKат для нeрeакционноспособных комплексов возрастает быстрее и более резко, чем для реакционноспособных. Напр., если a Кат f P /f PKaт равно с H5O2 + для реакционноспособных комплексов и равно h 0 , либо h 0 a H2O для нереакционноспособных. В водных растворах слабых кислот каталитич. активность проявляют ионы и недиссоциир. молекулы кислоты. Взаимосвязь между каталитич. активностью слабых кислот и их константами диссоциации в некоторых случаях удается описать корреляц. ур-нием Брёнстеда. В сильно разбавленных водных растворах оснований ндблюдается постоянство k эфф /c OH - при варьировании с OH -. Обоснованных количеств. характеристик ионизирующей способности умеренно конц. и конц. растворов оснований пока нет. Добавление солей к водным растворам кислот и оснований существенно изменяет их каталитич. активность; в случае слабых кислот и оснований изменяется степень их диссоциации на ионы из-за наличия общего иона и увеличения ионной силы раствора. В случае сильных кислот изменяются величины h 0 , а НА , а Н2O , что приводит к увеличению относит, концентрации комплексов, образующихся из частиц Н 3 О + А - , НА, а также усилению реакц. способности ионов Н 5 О 2 + , например при гидролизе в условиях, когда a Кат f P /f PKат =с H2O + . В случае сильных оснований катионы металлов усиливают каталитич. действие сольватир. ионов ОН - . Лимитирующие стадии катализируемых кислотами и основаниями реакций часто осуществляются по синхронным механизмам. В неводных растворителях кислоты и основания образуют преим. мол. комплексы. Каталитич. действие апротонных кислот обусловлено образованием из них и реагентов мол. реакционно-способных комплексов. Катиониты в Н-форме и нанесенные кислотные катализаторы обладают каталитич. активностью главным образом из-за наличия жидкой фазы, содержащей кислоту. Нанесенные кислотные катализаторы готовят нанесением на носитель (силикофосфат SiO 2 .Р 2 О 5 , силикагель, кварц, фосфаты) разл. жидких кислот, главным образом фосфорной. Каталитич. активность таких катализаторов зависит от кол-ва нанесенной кислоты на 1 г носителя и ее концентрации в жидкой фазе, зависящей от равновесного давления водяного пара над катализатором. Для характеристики ионизирующих свойств нанесенных кислотных катализаторов можно использовать такие же ф-ции а Кaт f P /f PKaт , как и для растворов. Специфич. свойство практически всех гетерог. кислотно-основных катализаторов наличие на их пов-сти центров разл. силы (кислотной или основной), что обусловлено неоднородностью пов-сти твердых тел. Из-за наличия подобных центров образуются сильно поляризованные комплексы реагента с катализатором. Энергетически выгодными являются синхронно протекающие лимитирующие стадии. Так, крекинг парафинов на Аl 2 О 3 может протекать по схеме:
Реакции в растворах часто ускоряются в присутствии веществ, относящихся к разновидностям кислот и оснований, названных именами Аррениуса, Льюиса, Бренстеда.
По общепринятому определению, данному в 1884 г. Сванте Аррениусом, кислота – соединение, дающее при диссоциации в воде катионы Н + , а основание – анионы ОН – .
По Д. Бренстеду и Т. Лоури (1923 г.) кислота – вещество, являющееся донором протона, а основание – акцептором протона.
Кислоту и основание, связанные уравнением типа (7.1), называют сопряженными
кислота протон основание
Протон в растворе обычно соединяется с молекулой растворителя, например с водой (7.2):
Если молекулы растворителя не присоединяют и не отдают протоны, то растворенное вещество не проявляет ни кислотных, ни основных свойств, а сам растворитель называют апротонным.
Раствор кислоты в воде – пример системы, состоящей из двух пар сопряженных кислот и оснований, находящихся в равновесии (7.3):
кислота основание кислота основание
По Гилберту Льюису (1938 г.) кислота – это акцептор неподеленной пары электронов, а основание – вещество, являющееся донором неподелённой пары электронов, которая может быть использована для образования устойчивой электронной конфигурации другого атома. Например, в реакции (7.4):
где H2O – основание, так как обладает свободной электронной парой, а SO3 – кислота, так как использует эту электронную пару.
Теория Льюиса гораздо шире, чем предыдущие теории, но имеет некоторые недостатки:
– сомнительно толкование протонных кислот, таких как H2SO4, НСl и других, так как они не могут присоединять электронную пару с образованием ковалентных связей;
– несостоятельность в определении силы кислот, поскольку по Льюису, это зависит от особенностей реакции.
Каталитические реакции, ускоряемые кислотами и основаниями, представлены широким перечнем химико-технологических процессов, таких как алкилирование парафинов и аренов олефинами, гидратация алкенов и алкинов, этерификация и гидролиз сложных эфиров, полимеризация олефинов в газовой фазе. Реакции, катализируемые кислотами и основаниями, можно разделить на три вида:
– специфический кислотный или основной катализ,
– общий кислотный или основной катализ,
– электрофильный или нуклеофильный катализ.
7.1.1. Специфический кислотный катализ
В этом случае катализаторами являются кислоты Аррениуса (ионы Н3О + ), и реакция идет по схеме (7.5):
где S – молекула субстрата,
P – молекула продукта.
Первая стадия – внедрение протона в реагирующую часть молекулы субстрата – протекает быстро. Вторая стадия, в которой образовавшийся катион SН + отщепляет протон с образованием продукта, протекает медленно и является лимитирующей стадией процесса.
По этому механизму протекают реакции гидролиза сложных эфиров и ацеталей, гидратации ненасыщенных альдегидов, дегидратации третичных спиртов, кетоенольной изомеризации.
7.1.2. Специфический основной катализ
Катализаторами здесь являются основания Аррениуса – гидроксид-ионы ОН – . Реакция протекает по схеме (7.6):
SH + ОН – ↔ S – + H2O → S * + OH – → P + OH – , (7.6)
где S * – новое промежуточное соединение, причем первая стадия быстрая, а вторая – медленная, то есть лимитирующая весь процесс.
По этому механизму протекают реакции гидролиза сложных эфиров, гидратации альдегидов, альдольной конденсации, реакции, включающие перегруппировки Кляйзена, Михаэля, Перкина.
7.1.3. Общий кислотный катализ
Он отличается от специфического тем, что здесь катализаторами процесса служат кислоты Бренстеда (НА), то есть ион Н3О + не является донором процесса, и реакция идет по схеме (7.7):
S + HA → SH + + A – → P + HA. (7.7)
Здесь медленной стадией является образование иона SH + , то есть лимитирующей является первая стадия процесса.
По этому механизму протекают реакции кетоенольной изомеризации, присоединения к карбонильной и карбоксильной группе.
7.1.4. Общий основной катализ
Катализаторами процесса выступают основания Бренстеда (В), то есть акцептором протона не является анион OH – . Процесс описывается схемой (7.8):

SH + B → S – + BH + → S * + B → P + B. (7.8)
Самой медленной стадией является образование активного аниона.
По этому механизму идут реакции гидролиза эфиров, конденсации альдегидов.
7.1.5. Электрофильный катализ
В этом случае катализаторами служат кислоты Льюиса. Объяснение роли кислот Льюиса как катализаторов связывают с образованием ими за счет донорно-акцепторной связи промежуточного соединения с одним реагентом, которое более легко вступает в реакцию с молекулами второго реагента благодаря наличию областей с повышенной или пониженной электронной плотностью.
По этому механизму протекают реакции Фриделя–Крафтса, гидролиза сложных эфиров аминокислот в присутствии катионов переходных металлов.
7.1.6. Кинетика реакций кислотно-основного катализа
Рассмотрим наиболее общий случай, когда реакция катализируется одновременно кислотами и основаниями Аррениуса (Н3О + и ОН – ).
Механизм такой реакции можно представить уравнениями (7.9 – 7.12):

SH + H3O + HSH + + H2O (7.9)

SH + OH – S – + H2O (7.10)

SH + H2O HSH + + OH – (7.11)

SH + H2O S – + H3O + (7.12)
При этом реагент SH участвует одновременно во всех четырех реакциях, отвечающих разным типам катализа.
Скорость расхода реагента SH запишем следующим образом (7.13):

(7.13)


. (7.14)
Сделаем некоторые допущения:

1) , где k0– константа скорости реакции реагента с водой (некаталитическая стадия);
2) , где – константа скорости реакции, катализируемой протонами;
3) , где – константа скорости реакции, катализируемой ионами гидроксила.
Таким образом, кажущаяся константа скорости реакции примет вид (7.15):

. (7.15)

Так как , то выражение (7.15) можно записать в виде (7.16):

, (7.16)

где ,
С 0 – стандартная концентрация.
Уравнение (7.16) позволяет рассмотреть все случаи кислотно-основного катализа. Графики зависимости lg [kкаж] = f(pH) для каждого случая приведены на рисунке 7.1.
Наиболее общий случай (а) изображен линией, которая указывает на три различные области рН, характеризуемые прямыми отрезками, наклон которых к оси абсцисс составляет – 1; 0 и + 1. В зависимости от величины рН преобладает либо специфический кислотный, либо основной катализ, а в промежуточной области – каталитическое действие воды. По данному типу катализа протекает реакция мутаротации глюкозы.
В случае (см. рис. 7.1, линия b) каталитическое действие воды происходит очень медленно по сравнению с реакциями, катализируемыми ионами Н + и ОН – . Существует два отрезка прямой (горизонтальный участок кривой отсутствует), которые пересекаются в точке с минимальным значением kкаж. Этому типу катализа отвечает реакция галогенирования ацетона.
Кривая с на рисунке 7.1 соответствует реакциям, катализируемым только ионами ОН – в растворах, когда рН > 7.
Кривая d представляет случай, когда реакция всегда катализируется ионами ОН – .
Кривая e описывает реакцию, катализируемую ионами Н + в растворах, когда рН имеет достаточно малые значения.
Кривая f характеризует пример, в котором реакция всегда катализируется ионами Н + .

Рис. 7.1. Логарифмическая зависимость кажущейся константы
скорости реакций, катализируемых кислотами и/или основаниями
Реакции в растворах часто ускоряются в присутствии веществ, относящихся к разновидностям кислот и оснований, названных именами Аррениуса, Льюиса, Бренстеда.
По общепринятому определению, данному в 1884 г. Сванте Аррениусом, кислота – соединение, дающее при диссоциации в воде катионы Н + , а основание – анионы ОН – .
По Д. Бренстеду и Т. Лоури (1923 г.) кислота – вещество, являющееся донором протона, а основание – акцептором протона.
Кислоту и основание, связанные уравнением типа (7.1), называют сопряженными
кислота протон основание
Протон в растворе обычно соединяется с молекулой растворителя, например с водой (7.2):
Если молекулы растворителя не присоединяют и не отдают протоны, то растворенное вещество не проявляет ни кислотных, ни основных свойств, а сам растворитель называют апротонным.
Раствор кислоты в воде – пример системы, состоящей из двух пар сопряженных кислот и оснований, находящихся в равновесии (7.3):
кислота основание кислота основание
По Гилберту Льюису (1938 г.) кислота – это акцептор неподеленной пары электронов, а основание – вещество, являющееся донором неподелённой пары электронов, которая может быть использована для образования устойчивой электронной конфигурации другого атома. Например, в реакции (7.4):
где H2O – основание, так как обладает свободной электронной парой, а SO3 – кислота, так как использует эту электронную пару.
Теория Льюиса гораздо шире, чем предыдущие теории, но имеет некоторые недостатки:
– сомнительно толкование протонных кислот, таких как H2SO4, НСl и других, так как они не могут присоединять электронную пару с образованием ковалентных связей;
– несостоятельность в определении силы кислот, поскольку по Льюису, это зависит от особенностей реакции.
Каталитические реакции, ускоряемые кислотами и основаниями, представлены широким перечнем химико-технологических процессов, таких как алкилирование парафинов и аренов олефинами, гидратация алкенов и алкинов, этерификация и гидролиз сложных эфиров, полимеризация олефинов в газовой фазе. Реакции, катализируемые кислотами и основаниями, можно разделить на три вида:
– специфический кислотный или основной катализ,
– общий кислотный или основной катализ,
– электрофильный или нуклеофильный катализ.
7.1.1. Специфический кислотный катализ
В этом случае катализаторами являются кислоты Аррениуса (ионы Н3О + ), и реакция идет по схеме (7.5):
где S – молекула субстрата,
P – молекула продукта.
Первая стадия – внедрение протона в реагирующую часть молекулы субстрата – протекает быстро. Вторая стадия, в которой образовавшийся катион SН + отщепляет протон с образованием продукта, протекает медленно и является лимитирующей стадией процесса.
По этому механизму протекают реакции гидролиза сложных эфиров и ацеталей, гидратации ненасыщенных альдегидов, дегидратации третичных спиртов, кетоенольной изомеризации.
7.1.2. Специфический основной катализ
Катализаторами здесь являются основания Аррениуса – гидроксид-ионы ОН – . Реакция протекает по схеме (7.6):
SH + ОН – ↔ S – + H2O → S * + OH – → P + OH – , (7.6)
где S * – новое промежуточное соединение, причем первая стадия быстрая, а вторая – медленная, то есть лимитирующая весь процесс.
По этому механизму протекают реакции гидролиза сложных эфиров, гидратации альдегидов, альдольной конденсации, реакции, включающие перегруппировки Кляйзена, Михаэля, Перкина.
7.1.3. Общий кислотный катализ
Он отличается от специфического тем, что здесь катализаторами процесса служат кислоты Бренстеда (НА), то есть ион Н3О + не является донором процесса, и реакция идет по схеме (7.7):
S + HA → SH + + A – → P + HA. (7.7)
Здесь медленной стадией является образование иона SH + , то есть лимитирующей является первая стадия процесса.
По этому механизму протекают реакции кетоенольной изомеризации, присоединения к карбонильной и карбоксильной группе.
7.1.4. Общий основной катализ
Катализаторами процесса выступают основания Бренстеда (В), то есть акцептором протона не является анион OH – . Процесс описывается схемой (7.8):
SH + B → S – + BH + → S * + B → P + B. (7.8)
Самой медленной стадией является образование активного аниона.
По этому механизму идут реакции гидролиза эфиров, конденсации альдегидов.
7.1.5. Электрофильный катализ
В этом случае катализаторами служат кислоты Льюиса. Объяснение роли кислот Льюиса как катализаторов связывают с образованием ими за счет донорно-акцепторной связи промежуточного соединения с одним реагентом, которое более легко вступает в реакцию с молекулами второго реагента благодаря наличию областей с повышенной или пониженной электронной плотностью.
По этому механизму протекают реакции Фриделя–Крафтса, гидролиза сложных эфиров аминокислот в присутствии катионов переходных металлов.
7.1.6. Кинетика реакций кислотно-основного катализа
Рассмотрим наиболее общий случай, когда реакция катализируется одновременно кислотами и основаниями Аррениуса (Н3О + и ОН – ).
Механизм такой реакции можно представить уравнениями (7.9 – 7.12):
SH + H3O + HSH + + H2O (7.9)
SH + OH – S – + H2O (7.10)
SH + H2O HSH + + OH – (7.11)
SH + H2O S – + H3O + (7.12)
При этом реагент SH участвует одновременно во всех четырех реакциях, отвечающих разным типам катализа.
Скорость расхода реагента SH запишем следующим образом (7.13):
(7.13)
. (7.14)
Сделаем некоторые допущения:
1) , где k0– константа скорости реакции реагента с водой (некаталитическая стадия);
2) , где – константа скорости реакции, катализируемой протонами;
3) , где – константа скорости реакции, катализируемой ионами гидроксила.
Таким образом, кажущаяся константа скорости реакции примет вид (7.15):
. (7.15)
Так как , то выражение (7.15) можно записать в виде (7.16):
, (7.16)
где ,
С 0 – стандартная концентрация.
Уравнение (7.16) позволяет рассмотреть все случаи кислотно-основного катализа. Графики зависимости lg [kкаж] = f(pH) для каждого случая приведены на рисунке 7.1.
Наиболее общий случай (а) изображен линией, которая указывает на три различные области рН, характеризуемые прямыми отрезками, наклон которых к оси абсцисс составляет – 1; 0 и + 1. В зависимости от величины рН преобладает либо специфический кислотный, либо основной катализ, а в промежуточной области – каталитическое действие воды. По данному типу катализа протекает реакция мутаротации глюкозы.
В случае (см. рис. 7.1, линия b) каталитическое действие воды происходит очень медленно по сравнению с реакциями, катализируемыми ионами Н + и ОН – . Существует два отрезка прямой (горизонтальный участок кривой отсутствует), которые пересекаются в точке с минимальным значением kкаж. Этому типу катализа отвечает реакция галогенирования ацетона.
Кривая с на рисунке 7.1 соответствует реакциям, катализируемым только ионами ОН – в растворах, когда рН > 7.
Кривая d представляет случай, когда реакция всегда катализируется ионами ОН – .
Кривая e описывает реакцию, катализируемую ионами Н + в растворах, когда рН имеет достаточно малые значения.
Кривая f характеризует пример, в котором реакция всегда катализируется ионами Н + .
Кислотность и основность соединений определяет их реакционную способность.
Согласно определениям Бренстеда и Лоури, кислота -- это вещество, отдающее протон, а основание -- вещество, которое протон присоединяет. Донорами и акцепторами протонов могут выступать как электронейтральные молекулы, так и ионы. Сопряженные пары кислот и оснований изображает реакция:
где $AH$ -- кислота;
Свойство кислот отдавать протон характеризуется константой кислотности $pK_a$. в водных растворах ее значение для протонных кислот лежит в широких пределах.
Примеры кислот и сопряженных с ними оснований: $N^+$, $NH_3$; $H_2O$, $OH^-$; $HSO_4^-$, $S^.$
По определению Льюиса кислотой является любое соединение, способное присоединить электронную пару, а основанием -- соединение, способное электронную пару отдать. Тогда все электронодефицитные соединения будут являться кислотами $(AlCl_3$, $BF_3$, $S_nCl_4)$ или основаниями (фосфины, амины, сульфиды, карбанионы).
Кислотно -- основный катализ вызван протолитической реакцией между кислотой и основанием и катализатором. Основная его особенность -- переход протонов от катализатора к реагенту и обратно.
Так, при кислотном катализе протон переходит от катализатора к молекуле реагирующего вещества. При основном катализе первоначально катализатор служит акцептором протона или донором аниона по отношению к молекуле реагента.
С ростом константы диссоциации кислот и оснований активность катализаторов в кислотно -- основном взаимодействии возрастает.
По типу кислотно -- основного катализа протекают реакции
- гидратации;
- дегидратации;
- этерификации;
- гидролиза;
- поликонденсации и др.
Электрофильные и нуклеофильные реагенты
Электрофильность и нуклеофильность являются характеристиками реакционной способности веществ, которые участвуют в бимолекулярных гетеролитических реакциях. Одна из частиц, электрофил, выступает акцептором пары электронов, а другая -- нуклеофил, донором электронов.
Скорость гетеролитической реакции будет тем больше, чем более ярко выражены электрофильные и нуклеофильные свойства.
Готовые работы на аналогичную тему
Электрофильные свойства увеличиваются при отщеплении протона от реагента основанием. Электрофильные и нуклеофильные свойства способны изменяться при комплексообразовании реагента с кислотой Льюиса.
Роль кислот и оснований в реакциях кислотно -- основного типа как катализаторов заключается в том, что они способны образовывать с реагентом промежуточное соединение в результате донорно -- акцепторного взаимодействия. В молекуле промежуточного соединения появляются реакционные центры, имеющие повышенную или пониженную электронную плотность, что способствует облегчению взаимодействия центрам с электрофильными или нуклеофильными молекулами второго реагента.
Классификация каталитических реакций кислотно -- основного типа
Все реакции кислотно -- основного типа в зависимости от природы частиц, выполняющих функции катализатора делят на группы:
Специфический кислотный катализ. Содержит реакции, катализируемые ионами водорода.
Гидролиз сложных эфиров в присутствии минеральных кислот. Включает присоединение протона к карбоксильному кислороду в результате донорно -- акцепторного взаимодействия. Образовавшийся карбоний -- катион обладает высокой электрофильностью, поэтому способен реагировать с водой. В ходе нескольких последовательных стадий образуется спирт, кислота и протон, способный вновь вступать в реакции.
Специфический основный катализ. Включает реакции, катализируемые ионами гидроксила. Промежуточными соединениями выступают анионы, образующиеся в результате присоединения основания или в результате отрыва катализатором протона от реагента.
Альдольная конденсация в присутствии гидроксил -- ионов протекает в несколько этапов:
- отщепление протона и образование енолят -- иона.
- присоединение енолят -- иона к карбонильной группе другой молекулы.
- отщепление протона от растворителя, образование продукта реакции и регенерация катализатора.
Общий кислотно -- основный катализ. Включает реакции, катализируемые кислотами и основаниями Бренстеда. В активации молекулы реагента принимают участие электрофильные частицы и кислоты Льюиса, которые выступают в качестве протона.
Реакция Фриделя -- Крафтса (алкилирование ароматического кольца) катализируется хлоридом алюминия $AlCl_3$ в среде неполярного растворителя. Сначала образуется поляризованная молекула промежуточного соединения, возникает электрофильный реакционный центр. Этот центр вступает во взаимодействие с молекулой бензола с образованием $\sigma $- комплекса, один из атомов углерода переходит из состояния $sp^2$-гибридизации в $sp^3$-гибридизацию. Образуется продукт и катализатор.
Электрофильно -- нуклеофильный катализ. Содержит реакции катализируемые донорами или акцепторами электронных пар.
Гидролиз сложного эфира в присутствии имидазола -- типичный пример нуклеофильного катализа. Имидзол взаимодействует с эфиром и этим облегчает реакцию с водой. Молекулы промежуточного соединения сильно поляризуются и обладают большой реакционной способностью. В качестве нуклеофильных катализаторов могут выступать амины, ионы галогенов, некоторые анионы.
Зависимость константы скорости от функций кислотности
Реакции в органической химии могут катализироваться как кислотами, так и основаниями.
Экспериментально кислотно -- основный катализ легко распознать по кривым зависимости логарифма константы скорости от $pH$, $H_0$ и других функций кислотности. Частный случай такой зависимости: двухстадийная реакция. Одна стадия ингибируется при добавлении кислоты, а другая в это время ускоряется.

В реакции кетонов с гидроксиламином образование аминов будет замедляться в кислой среде. Это происходит из-за дезактивации гидроксиламина протонированием. Дегидратация, вторая стадия, катализируется кислотой и будет замедляться при высоких $pH$.
Тогда уравнение скорости между протонированной формой A и Y выразится как
Уравнение (13) отражает противоборствующее влияние кислотности среды на скорость реакции: 1-й член увеличивается с ростом кислотности, но 2-й уменьшается. Такой характер влияния обусловливает экстремальную зависимость скорости соответствующих реакций от кислотности среды. Подобные зависимости наблюдаются для кислотно-каталитических реакций карбонильных соединений с азотистыми основаниями.
Гетерогенный катализ важнейшая область катализа, имеющая широкое практическое приложение. Достаточно в этом плане отметить такие важные процессы как синтез аммиака
В то же время гетерогенно-каталитические реакции – сложные и многомерные физико-химические системы, представляющие большой научный интерес.
Преимущества гетерогенного катализа перед гомогенным состоят в малом расходе катализатора на единицу количества продукта, снижении или полном устранении токсичных сточных вод и расходов дополнительных реагентов на промывку реакционной массы. Это обусловлено тем, что катализатор и реакционная масса находятся в разных фазах.
По способу осуществления гетерогенно-каталитические процессы делятся на процессы со стационарным (неподвижным) катализатором, где он используется в виде достаточно крупных гранул (0,3 – 1 см) и на процессы с подвижным катализатором (плавающим, диспергированным или псевдоожиженным). В последнем случае катализатор применяют только в диспергированном виде, когда он способен перемешиваться под влиянием потока реакционной массы.
Каталитические процессы осуществляются в газовой или жидкой фазе.
В зависимости от типа механизма реакций, протекающих на гетерогенных катализаторах последние подразделяют на следующие группы:
1. Ионные, под влиянием которых протекают реакции по ионному механизму.
2. Электронные, катализирующие гомолитические реакции
3. Бифункциональные, совмещающие ионный и электронный катализ
К ионным катализаторам относятся следующие:
1. Кислотно-основные катализаторы
В группу этих катализаторов входят
а) оксиды некоторых металлов, например Al 2 O 3 , W 2 O 3 , ThO 2 .
б) нейтральные и кислые соли: Ca 3 ( PO 4 )2, CaHPO 4 , MgHPO 4 , а также природные и синтетические алюмосиликаты ( Al 2 O 3 ) m ( SiO 2 ) n ( H 2 O ) p .
в) Протонные и апротонные кислоты на носителях ( H 3 PO 4 на Al 2 O 3 или кизельгуре, BF 3 на Al 2 O 3 , гетерополикислоты) и ионообменные смолы.
2. Комплексообразующие соли переходных металлов на носителях: Cu 2 Cl 2 , HgCl 2 , PdCl 2 + CuCl 2 , CuC º C С u , NiCl 2 и другие.
Все катализаторы ионного типа являются изоляторами или ионными проводниками электрического тока. Наиболее распространены катализаторы кислотного типа, являющиеся протонными (бренстедовскими) или апротонными (льюисовскими) кислотами.
Гетерогенные катализаторы гомолитических реакций всегда являются электронными проводниками тока или полупроводниками, они включают следующие группы веществ:
1. Перехолные металлы I Б и VIII групп ( Cu , Ag , Fe , Ni , Co , Pt , Pd ) периодической системы Д. И. Менделеева.
2. Оксиды металлов ( MgO , ZnO , CuO , Fe 2 O 3 , Cr 2 O 3 , WO 3 , MoO 3 , V 2 O 5 ) сульфиды ( WS 3 , MoS 3 ) и смеси, содержащие один основной оксид с небольшими добавками других (модифицированные оксидные катализаторы)
3. Сложные оксидные и сульфидные катализаторы с соизмеримым соотношением компонентов, а также соли-полупроводники: хромиты ( CrO × Cr 2 O 3 , ZnO × Cr 2 O 3 ), вольфраматы ( CoO × WO 3 ), молибдаты ( Bi 2 O 3 × MoO 3 , NiS × MoS 3 ) ванадаты и другие.
Между ионными и электронными катализаторами не существует резкой границы, некоторые из них способны ускорять ионные и гомолитические реакции. Из индивидуальных веществ это особенно относится к оксидам, часть из которых катализирует ионные, а остальные – гомолитические реакции. Относительная активность оксидных катализаторов в ионных и гомолитических реакциях может быть проиллюстрирована на примере конкуренции двух реакций – ионной реакции дегидратации этанола и гомолитический реакции – его дегидрирования.
относительная доля продукта дегидратации, %
относительная доля продукта дегидратации, %
Из приведенных данных видно, что некоторые оксиды являются бифункциональными катализаторами. Бифунциональности катализаторов можно также достичь используя смеси оксидов разного типа. Лучшим примером является система ZnO на Al 2 O 3 , успешно применявшаяся для синтеза бутадиена из этанола, который представляет собой совмещенный процесс дегидратации, димеризации и дегидрирования. Другой пример бифункциональных катализаторов – металлы платиновой группы на носителях кислотного типа ( Al 2 O 3, алюмосиликаты), являются катализаторами каталитического риформина в котором протекают гомолитические реакции дегидрирования и ионные реакции изомеризации и расщепления.
Гетерогенные катализаторы должны удовлетворять следующим требованиям:
- высокая каталитическая активность
- достаточно большая селективность (избирательность) в отношении целевой реакции
- простота получения, обеспечивающая воспроизводимость всех свойств катализатора.
- высокая механическая прочность к сжатию, удару и истиранию
- достаточная стабильность всех свойств катализатора на протяжении его службы и способность к их восстановлению при том или ином методе регенерации.
- небольшие экономические затраты на катализатор при производстве единицы продукции.
Обеспечение этих требований достигается главным образом при разработке состава катализатора и способа его получения.
Гетерогенные катализаторы сравнительно редко применяются в индивидуальном виде и часто содержат различные добавки, получившие название модификаторов. Цели их введения разнообразны: повышение активности катализатора (промоторы), избирательности и стабильности работы, улучшение механических или структурных свойств. Фазовые и структурные модификации стабилизируют соответственно активную фазу твердого катализатора или пористую структуру его поверхности. Так, в меди-хромистых катализаторах гидрирования оксид хрома препятствует восстановдению оксида меди с превращением его в неактивную форму. Добавление уже 1% Al 2 O 3 к железному катализатору значительно увеличивает его поверхность, препятствуя спеканию и закрытию пор. Некоторые модификаторы существенно повышают стабильность работы катализатора.
В смешанных катализаторах, где компоненты находятся в соизмеримых количествах, могут образовываться новые более активные соединения, их твердые растворы в основном компоненте или многофазные системы, обладающие специфическим каталитическим действием. При этом свойства смешанного катализатора не являются простой суммой свойств его компонентов. Это приводит к заметному увеличению избирательности, активности и других свойств катализатора.
К числу модификаторов можно отнести и носители (трегеры), особенно часто применяемые в случае дорогостоящих металлических катализаторов ( Pt , Pd , Ni , Co ). Роль последнего состоит в повышении активной поверхности, увеличении термостойкости и механической прочности катализатора и т.д. В качестве последнего используют алюмосиликаты, Al 2 O 3 , Cr 2 O 3 , SiO 2 , активированный уголь, пемзу, кизельгур и другие природные и синтетические материалы.
Как правило, гетерогенно-каталитический процесс протекает через ряд последовательных, а иногда и последовательно параллельных стадий, существенно различающихся по своей природе. К таким процессам относятся:
1. диффузия реагентов из потока к внешней поверхности зерна катализатора.
2. диффузия реагентов к внутренней поверхности зерна катализатора (в поры)
3. адсорбция реагентов на поверхности катализатора
4. собственно химическая реакция
5. десорбция продуктов реакции с поверхности катализатора
6. диффузия продуктов с внутренней поверхности зерна катализатора
7. диффузия продуктов с внешней поверхности зерна катализатора в поток
Любая из этих стадий может оказаться лимитирующей и, следовательно, определять скорость процесса в целом. Таким образом, кинетические закономерности гетерогенно-каталитического процесса могут контролироваться закономерностями диффузии, адсорбции или химической реакции, а в пограничных случаях – их совокупностью. Это определяет специфику кинетики гетерогенного катализа по сравнению с кинетикой гомогенного катализа.
Принято различать кинетически- и диффузионно- контролируемые области протекания гетерогенно-каталитических процессов. В первых из них общая скорость процесса определяется скоростью химической реакции на поверхности, во вторых – диффузией реагентов или продуктов реакции. Кроме того, существуют области, контролируемые сорбцией реагентов или десорбцией продуктов.
Более детально различают следующие 5 основных областей:
1. Внешнедиффузионная область. В этой области скорость в целом определяется скоростью транспорта реагентов из потока к внешней поверхности зерна катализатора (или скоростью диффузии продуктов с внешней поверхности в поток)
2. Внутридиффузионная область. В этом случае скорость процесса лимитируется диффузией реагентов от внешней поверхности зерна катализатора к его внутренней поверхности (или наоборот, диффузией продуктов реакции из пор катализатора к его внешней поверхности)
3. Внешнекинетическая область. В этом случае скорость процесса лимитируется скоростью химической реакции на внешней поверхности зерна катализатора. Это возможно, если скорость химической реакции на внешней поверхности значительно превосходит скорость внутренней диффузии (стадии 2 и 6), но значительно меньше скорости внешней диффузии.
4. Внутрикинетическая область. Скорость процесса определяется скоростью химической реакции, причем последняя протекает на внешней и внутренней поверхности зерна катализатора. Это возможно, когда химическая реакция идет значительно медленнее внешней и внутренней диффузии.
5. Сорбционная область. Здесь скорость процесса определяется адсорбцией реагентов или десорбцией продуктов.
Строгие границы между этими областями отсутствуют, они перекрываются переходными областями, в которых сочетаются закономерности различных областей.
При экспериментальном исследовании и расчетах гетерогенно-каталитических процессов важно знать область, в которой протекает реакция, отчего зависит вид описывающих её кинетических уравнений. Так, при лимитировании скорости процесса химической реакцией на всей поверхности катализатора (внутрикинетическая область) скорость диффузии не играет роли, и результаты процесса не будут зависеть от размера зерен катализатора. Наоборот: во внешне- или внутридиффузионной области размер зерна играет большую роль, так как скорость диффузии на единицу массы катализатора зависит от величины внешней поверхности, которая определяет и диффузию в поры. Таким образом, проводя серию экспериментов с катализатором разного размера зерна и наблюдая за изменением конверсии, можно различить кинетическую и диффузионную области, а также определить размер зерна катализатора, необходимый для достижения кинетической области.
Тестом на различие внешне- и внутридиффузионной областей является зависимость скорости внешней диффузии от линейной скорости газового потока, тогда как внутренняя диффузия не зависит от этого параметра. Линейную скорость потока можно варьировать, меняя объем катализатора, соблюдая при этом постоянство времени контакта.
Легко убедиться, что такое требование можно соблюсти при условии пропорционального изменения объема υ линейной скорости n , связанной с объемной скоростью потока соотношением
Изменение или постоянство степени конверсии в таких опытах свидетельствует о наличии или отсутствии внешнедиффузионного торможения.
При исследовании гетерогенно-каталитических реакций наиболее достоверные данные получаются в проточно-циркуляционных установках, хотя часто используют метод исследования в потоке.
Во избежание нарушений в режиме потока реагентов диаметр каталитической ячейки или трубы должен быть равным не менее 6-7 диаметров зерна катализатора.
Обычно при работе катализаторов происходит постепенное изменение их активности и селективности. Поэтому кинетический эксперимент можно ставить лишь в период их стационарных свойств, который наступает после более или менее длительного холостого пробега и заканчивается при наличии признаков заметной дезактивации катализаторов.
Кинетические эксперименты проводят при варьировании начальных концентраций (парциальных давлений) реагентов и продуктов, а также условного времени контакта.
При кинетическом анализе реакций на поверхности катализатора руководствуются законом действующих поверхностей, согласно которому скорость химической реакции пропорциональна поверхностной концентрации реагентов, участвующих в акте химического взаимодействия.
В случае мономолекулярных реакций, например A ® B , акт химического взаимодействия можем представлять собой превращение вещества, сорбированного на активном центре поверхности, или его взаимодействия с соседним свободным центром поверхности. Эти двум механизмам соответствуют кинетические уравнения
В случае бимолекулярных реакций, например A + Y ® B , возможны также два механизма. Согласно одному из них два реагента адсорбируются на разных активных центрах поверхности и взаимодействуют между собой, если эти центры находятся в достаточной близости друг от друга (механизм Хиншельвуда). Такому механизму соответствует следующее кинетическое уравнение
Согласно другому механизму на поверхности сорбируется один из реагентов, активирования которого достаточно для взаимодействия с налетающей из объема молекулой второго реагента (“ударный” механизм Ридила). Такому механизму соответствует кинетическое уравнение
Неэлементарные реакции состоят из ряда элементарных стадий, составляющих их механизм. Кинетика таких реакций определяется последовательностью элементарных стадий, их характером (обратимые, необратимые), природой реагентов, интермедиатов и продуктов реакции. При кинетическом анализе неэлементарных реакций возникает задача определения концентраций интермедиатов, играющих ключевую роль в образовании продуктов или расходовании реагентов. В качестве инструмента такого определения используется принцип квазистационарных концентраций Боденштейна – Семенова. Согласно этому принципу скорость изменения концентраций нестабильных интермедиатов пренебрежимо мала по сравнению со скоростью изменения концентраций реагентов и продуктов реакции и её можно считать равной нулю. Применение принципа стационарных концентраций к неэлементарным реакциям, протекающим по сложному механизму, позволяет исключить из кинетического описания процессов неизвестные концентрации интермедиатов и получить одно или некоторый минимум дифференциальных уравнений скорости, выраженных через подлежащие измерению концентрации реагентов и продуктов реакции.
КИСЛOТНО-ОСНОВНOЙ КАТАЛИЗ
КИСЛOТНО-ОСНОВНOЙ КАТАЛИЗ (кислотно-основный катализ), ускорение хим. р-ций в присут. к-т и оснований. В качестве катализаторов используют: в гомог. кислотном катализе протонные к-ты (H 2 SO 4 , HCl, Н 3 РО 4 , CH 3 C 6 H 4 SO 2 OH и др.) в воде и водно-орг. р-рителях, апротонные к-ты (АlС1 3 , BF 3 , SnCl 4 и др.) в неводных р-рителях, сверхкислоты (HF SbF 5 , HSO 3 F SbF 5 и др.) в неводных р-рителях; в гетерог. кислотном катализе прир. глины, аморфные и кристаллич. алюмосиликаты, фосфорную и полифосфорные к-ты, нанесенные на носитель, катиониты; в гомог. основном катализе оксиды щелочных металлов, амины в воде, орг. и водно-орг. р-рителях; в гетерог основном катализе - оксиды металлов (CaO, MgO и др.). При кислотно-основном катализе в большинстве случаев из реагентов и катализатора в равновесных стадиях образуются реакционноспособные (а также нереакционноспособные) комплексы разл. состава, к-рые являются ионизир. формой реагента. В лимитирующих стадиях комплексы превращ. в продукты р-ции. В случае р-ции с одним реагентом (Р) эти стадии выражаются ур-нием:
где Кат-разл. формы катализатора: недиссоциир. к-ты или основания, ионы или ионные пары, образующиеся при их ионизации; P 1 Кат-комплексы; К i - константа равновесия образования комплексов Р 1 Кат; k ист - элементарная константа скорости превращ. комплексов в продукты. В случае р-ции двух реагентов (P 1 +Р 2 : Продукты; Р 2 +Р 2 : Продукты) реакционноспособный комплекс образуется, как правило, из комплекса одного реагента с несвязанным в комплекс др. реагентом:
где К к - константа равновесия образования комплексов Р 1 Kат Р 2 . Константы равновесия K i и К к выражают ур-ниями:
где (здесь и далее) а, с, f-соотв. термодинамич. активность, концентрация и коэф. активности реагирующих частиц; a Kат f P1 /f P1Кат - ф-ция, характеризующая способность среды переводить реагенты в комплексы РКат. При варьировании в широких пределах с Кат реализуются (особенно в р-рах) неск. маршрутов р-ции - хим. превращений, приводящих к образованию одних и тех же продуктов через реакционноспособные комплексы (РК) разл. состава. В р-ре с постоянной с Кат скорость р-ции по каждому маршруту W i выражается ур-нием:
где (k ист ) i - элементарная константа скорости по i-му маршруту, а* и f* - относятся к активир. комплексу. Из опытных данных следует, что множитель f РК /f* не изменяется при варьировании с Кат и его приравнивают к единице. В р-ре с постоянной с Кат наблюдаемая скорость р-ции
Экспериментально определяют эффективную константу скорости k эфф , характеризующую скорость р-ции при постоянной т-ре и наличии катализатора определенного состава. В ур-ниях для связи f эфф со св-вами катализатора учитывают все его комплексы с реагентом: реакционноспособные (РКат) РК и нереакционноспособные (РКат) НРК . Напр., при кислотном гидролизе амидов, анилидов ионизированная по атому азота форма является реакционноспособной, ионизированная по карбонильной группе - либо намного менее реакционноспособна, либо нереакционноспособна. Для описания влияния ионизирующих св-в среды на k эфф совместно решают ур-ние материального баланса для текущих концентраций реагентов и ур-ния (3) и (5) в случае р-ции одного реагента и дополнительно ур-ние (4) для р-ции двух реагентов. напр. для р-ции, протекающей по схеме (2):
В случае р-ции двух реагентов, если один (Р 2 ) является компонентом р-рителя (напр., Н 2 О при гидролизе в водных р-рах), при условии с р2 >>е р1 вычисляют эффективную константу скорости первого порядка, включающую термодинамич. активность р-рителя. В р-циях Р х с Р 2 в р-рах к-т и оснований реакционноспособный комплекс состава P 1 KaтP 2 может получаться и в лимитирующей стадии из ионизир. формы одного реагента и неионизир. формы другого:
В этом случае для описания влияния ионизирующих св-в среды на k эфф пригодно ур-ние (6), при замене величины (f P2 с P2 )/(К к f P1КатP2 ) на термодинамич. активность растворителя. Поскольку крайне редко удается установить прямыми физ. методами состав и концентрацию промежут. комплексов, в настоящее время основным методом установления их состава, детального механизма р-ции и вычисления величин К i и К к является кинетич. метод, основанный на применении ур-ний типа (6) для объяснения наблюдаемой зависимости k эфф от ионизирующей способности среды. Для этого измеряют k эфф в широком диапазоне с Кат в р-ре и подбирают ф-ции а Кат f P /f PКат для описания влияния среды на k эфф . наиб. исследованы каталитич. св-ва сильных к-т в водных р-рах. Ионизирующая способность таких р-ров обусловлена образованием комплексов реагента: 1) с сольватированным протоном и 2) с недиссоциированной к-той. К первым относятся протонир. форма реагента и ее комплексы с р-рителем. В этом случае а Кат f P /f PKат выражают (для водных р-ров) ф-циями h 0 =a H3O+ f P /f PH+ , h 0 a H2O и h 0 a 2 Н2O , где h 0 - измеренная индикаторным методом кислотность среды, f РН+ -коэф. активности протонир. формы реагента. Ко вторым относятся комплексы реагента с к-той (НА) и ее гидратами. При этом a Кат f Р /f РКат кат принимает значения а НА , a НА a Н2O и др. При гидролизе нек-рых карбонильных соед. a Кат f P /f РКат =c H5O2 + Однозначную информацию о составе реакционноспособных комплексов, как правило, получают на основании данных по изменению k эфф в р-рителе постоянного состава при варьировании с Кат в широких пределах в условиях малой степени связывания реагента в комплексы. наиб. простые зависимости получают для р-ции одного реагента, когда он равновесно связывается только в один реакционноспособный комплекс. В таком случае ур-ние (4) упрощается:
Опытным путем установлено, что в водных р-рах сильных к-т k ист не зависит от с Кат . К i , по определению, не должно зависеть от с Кат . Вопрос о влиянии состава смешанного р-рителя на k ист пока достаточно не исследован. В условиях, когда K i и (a Кат f Р )/f РКат соизмеримы, по ур-нию (8) вычисляют раздельно k ист и K i . При а Кат f Р /f РКат >> K i величина k эфф =k ист ; при k Кат f P /f РКат i имеет место соотношение:
Ниже приведен пример, раскрывающий природу каталитич. действия водных р-ров к-т в условиях, когда реакционноспособный комплекс образуется и превращ. в продукты согласно схеме (1) при условии, что а Кат f P /f РКат i . Ионизирующая способность среды из-за превращ. реагента в протонир. форму и ее комплексы с Н 2 О проявляется при наличии зависимостей К эфф /h 0 =const, k эфф (h 0 a H2О )=const k эфф /(h 0 a 2 H2O )=const. Если в р-ции k эфф /h 0 =const (lgk эфф +H 0 =const, где H 0 =-lgh 0 - ф-ция кислотности Гаммета), то a кат f P /f Кат =h 0 и реакционноспособным комплексом является протонир. форма реагента РН 3 О + . Такая закономерность имеет место при проведении пинаколиновой перегруппировки в конц. H 2 SO 4 :
При варьировании с Kaт значение k эфф /(h 0 а H2O ) постоянно в р-циях дегидратации, напр. 2-фенил-2-пропанола в серной к-те, 2-метил-2-бутанола в азотной к-те. Следовательно, а Kат f P /f PKaт равно h 0 а H2O и реакционноспособным является комплекс молекулы воды с протонир. формой реагента. Имеется и пример постоянства k эфф /(h 0 a 2 H2O ), что наблюдается в р-ции изотопного обмена атома кислорода в СН 3 ОН, протекающего в серной и хлорной к-тах. В этом случае а Kaт /f P /f РКaт =h 0 a 2 H2O и реакционноспособным является симметричный комплекс:
Ф-ции h 0 , h 0 а H2O и h 0 а 2 H2O стандартизованы к воде, и в сильно разбавленных р-рах сильных к-т они численно равны концентрации ионов Н 5 О + 2 в моль/л. В таких р-рах для приведенных выше р-ций при варьировании с Kaт должно соблюдаться постоянство значений k эфф /c H2O + . При гидролизе нек-рых амидов, анилидов, сложных эфиров k эфф /c H2O + постоянно не только в разбавленных, но и в конц. водный р-рах НСlO 4 , H 2 SO 4 . Если гидролиз протекает по схеме (1), то реакционноспособный комплекс образуется из реагента и иона Н 5 О 2 + , а выраженная в концентрациях константа равновесия K H5O2 + ; не изменяется при варьировании с Кат :
Не исключено, что k эфф /c H2O + =const из-за того, что р-ция протекает по схеме (7) Ионизирующая способность среды из-за комплексообразования реагента с молекулами недиссоциир. сильной к-ты и ее гидратом проявляется при наличии зависимостей k эфф /a HA =const, k эфф /(a HA a H2O )=const и др. В первом случае a Kaт =const и реакци онноспособный комплекс имеет состав РИА; такая закономерность, напр., реализуется при дегидратации b -фенил- b -гидроксипропионовой к-ты и 2-(4-мето-ксифенил)-2-пропанола в серной к-те. Во втором случае а кат f P /f PKат =а HА а H2O и комплекс имеет состав РНАН 2 О. Гидрат НА . Н 2 О можно рассматривать и как ионную пару Н 3 О + А - . Каталитич. активность гидратов сильных к-т проявляется в р-циях дегидратации 1-фенилэтанола в хлорной к-те, гидратации цис-коричной к-ты и рацемизации b -фенил- b -гидроксипропионовой к-ты в серной к-те. Согласно ур-нию (6), должен наблюдаться максимум k эфф в зависимости от с Кат , если наряду с реакционноспособными образуются и нереакционноспособные комплексы и при увеличении с Кат значение a Кат /f P /f PKат для нeрeакционноспособных комплексов возрастает быстрее и более резко, чем для реакционноспособных. Напр., если a Кат f P /f PKaт равно с H5O2 + для реакционноспособных комплексов и равно h 0 , либо h 0 a H2O для нереакционноспособных. В водных р-рах слабых к-т каталитич. активность проявляют ионы и недиссоциир. молекулы к-ты. Взаимосвязь между каталитич. активностью слабых к-т и их константами диссоциации в нек-рых случаях удается описать корреляц. ур-нием Брёнстеда. В сильно разбавленных водных р-рах оснований ндблюдается постоянство k эфф /c OH - при варьировании с OH -. Обоснованных количеств. характеристик ионизирующей способности умеренно конц. и конц. р-ров оснований пока нет. Добавление солей к водным р-рам к-т и оснований существенно изменяет их каталитич. активность; в случае слабых к-т и оснований изменяется степень их диссоциации на ионы из-за наличия общего иона и увеличения ионной силы р-ра. В случае сильных к-т изменяются величины h 0 , а НА , а Н2O , что приводит к увеличению относит, концентрации комплексов, образующихся из частиц Н 3 О + А - , НА, а также усилению реакц. способности ионов Н 5 О 2 + , напр. при гидролизе в условиях, когда a Кат f P /f PKат =с H2O + . В случае сильных оснований катионы металлов усиливают каталитич. действие сольватир. ионов ОН - . Лимитирующие стадии катализируемых к-тами и основаниями р-ций часто осуществляются по синхронным механизмам. В неводных р-рителях к-ты и основания образуют преим. мол. комплексы. Каталитич. действие апротонных к-т обусловлено образованием из них и реагентов мол. реакционно-способных комплексов. Катиониты в Н-форме и нанесенные кислотные катализаторы обладают каталитич. активностью гл. обр. из-за наличия жидкой фазы, содержащей к-ту. Нанесенные кислотные катализаторы готовят нанесением на носитель (силикофосфат SiO 2 . Р 2 О 5 , силикагель, кварц, фосфаты) разл. жидких к-т, гл. обр. фосфорной. Каталитич. активность таких катализаторов зависит от кол-ва нанесенной к-ты на 1 г носителя и ее концентрации в жидкой фазе, зависящей от равновесного давления водяного пара над катализатором. Для характеристики ионизирующих св-в нанесенных кислотных катализаторов можно использовать такие же ф-ции а Кaт f P /f PKaт , как и для р-ров. Специфич. св-во практически всех гетерог. кислотно-основных катализаторов наличие на их пов-сти центров разл. силы (кислотной или основной), что обусловлено неоднородностью пов-сти твердых тел. Из-за наличия подобных центров образуются сильно поляризованные комплексы реагента с катализатором. Энергетически выгодными являются синхронно протекающие лимитирующие стадии. Так, крекинг парафинов на Аl 2 О 3 может протекать по схеме:
Лит. Гам мет Л , Основы физической органической химии, пер. с англ., М., 1972; Танабе К . Твердые кислоты и основания, пер. с англ., М., 1973, Белл Р , Протон в химии, пер. с англ., М., 1977; Виниик М И , "Кинетика и катализ", 1980. т 21. в. I.e. 136 58. Казанский В Б . там же. 1987. т 28. в. I, с. 47 60 МИ Винник
Читайте также:
- Опишите технологию создания и форматирования текста с помощью текстового процессора word кратко
- Психолог в школе приемных родителей
- Методика обучения грамоте в старшей группе детского сада
- Биография чарушина для детей начальной школы кратко презентация
- История развития детского движения за рубежом кратко