
Доклад апоптоз его значение в физиологических и патологических процессах
Обновлено: 05.07.2024
Концепция апоптоза как явления "запрограммированной" гибели клеток приобретает в последнее время все больше фактов и вариантов ее приложения к базовым вопросам современной медицины.
Термин "апоптоз" впервые был предложен в 1972 г. для обозначения генетически обусловленного процесса разрушения клетки и характеризуемого ее сжатием, агрегацией хроматина и деструкцией клеточного ядра. Апоптоз рассматривается как естественный биологический механизм, который способствует ликвидации "ненужных" клеток и тканей. Биологическая "цель" этого явления состоит в удалении нежелательных клеток в процессе индивидуального развития, при защитных реакциях, старении. Физиологическое назначение апоптоза состоит в селекции разновидностей и качества клеток внутри популяции, в том числе удалении клеток с генетическими дефектами, а также поддержании численности клеток тканевой популяции на функционально необходимом уровне.
Эмбриональное и постэмбриональное развитие мозга сопровождается изменениями числа, структуры и функциональных "качеств" нервных клеток. Соотношение процессов возникновения новых структур и ликвидации "ненужных" клеток регулируется апоптическими процессами. Закономерный характер апоптозных реакций прослеживается как на субклеточном уровне (митоптоз - ликвидация митохондрий), так и в целом организме - в процессе индивидуального развития, когда наблюдаются регрессия рудиментарных органов, перестройка клеточного пула при росте и дифференцировке тканей.
В "нормальной" ткани механизмы апоптоза пребывают под регулируемым генетическим контролем. Как было недавно установлено, ограничение апоптозного механизма в нейронах эмбрионального мозга, регулируемое нейротрофическими ростовыми факторами и особыми белками, ведет к увеличению числа синапс - образующих клеток нейрональной популяции в целом.
Однако помимо общебиологического (общефизиологического) значения это явление оказывается значимым в процессах онкообразования, аутоиммунных патологиях, вирусных инфекциях, сердечно-сосудистых заболеваниях. Это представление получает также все большее число иллюстраций при изучении неврологических патологий. За последние пять лет, согласно информации Medline, по проблеме апоптоза опубликовано около 40 тыс. экспериментальных и клинических статей.
НЕЙРОАПОПТОЗ
Биохимические и морфоцитологические признаки апоптоза выявляются в большом спектре экспериментальных нейродегенеративных расстройств: транзиторной церебральной ишемии, вызванной окклюзией церебральной артерии; интрацеребральной геморрагии; на моделях эпилептогенных судорог, локальном термическом повреждении мозга. Апоптоз включается в патологии любых проявлений ишемических и травматических повреждений нервной ткани, включая нарушения спинного мозга, деменциальные изменения, связанные с развитием болезней Альцгеймера, Паркинсона, сенильной деменцией, врожденными патологиями мозга и др. Таким образом, можно говорить о нейроапоптозе как новом патобиохимическом механизме нейродегенеративных расстройств широкого спектра.
Основное для понимания апоптоза как принципиально нового морфо-биохимического процесса, отличного от некроза, - включение специализированных биохимических механизмов, в результате которых происходит разрыв молекулы ДНК и уничтожение белоксинтезирующих структур. Апоптоз развивается как каскадный процесс, который сопровождается активацией (индукцией образования) специфических про- или антиапоптических белков, а также особых протеолитических ферментов - каспаз.
МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ АПОПТОЗА
1. Нарушение энергетического потенциала митохондрий.
2. Образование активных форм кислорода (АФК).
3. Запуск "специализированных" биохимических систем.
4. Деструкция ДНК.
5. Морфологическая гибель клетки, ткани, организма.
Среди факторов запуска апоптоза следует отметить образование активных форм кислорода как "извращенного" пути окислительного метаболизма в клетке. Считается, что повреждения, развивающиеся в результате аноксии или ишемии ткани любого уровня, обязаны в первую очередь образованию АФК. Первичным источником АФК оказываются митохондрии, которые играют ключевую роль в энергетическом обеспечении клетки. Ныне существует обобщенное клиническое понятие "митохондриальная патология": повреждение мембран митохондрий ведет к образованию супероксидных радикалов, которые, реагируя с NO, образуют пероксинитриты, "повреждающие" молекулы АФК.
В точных биохимических исследованиях было установлено, что нарушение "нормальной" аккумуляции Са++ митохондриями, высвобождение апоптогенных белков из поврежденных митохондрий в цитозоль служат механизмами, ответственными за индукцию апоптоза. В этом контексте существенна роль одного из нейротрофических факторов - фактора некроза опухоли (TNF-(), с которым связаны открытие пор в митохондриях, последующий разрыв их мембраны и высвобождение проапоптических белков в цитозоль клетки.
Факторы, последовательно включающиеся в апоптоз:
1.Гиперпродукция экзайтоксических (excitoxic - возбуждающие) аминокислот и длительная активация глутаматных рецепторов, выраженные при нарушении функции нервных клеток вследствие возрастных нейродеструктивных процессов, болезни Альцгеймера, паркинсонизме, при остром нарушении мозгового кровообращения.
2.Образование АФК, которые стимулируют синтез провоспалительных цитокинов, включающихся в каскад апоптических процессов.
3. Экспрессия группы рецепторов и проапоптических белков (FAS, APO-1 и др.). Один из наиболее значимых в этой группе - белок p53.
4. Включение каскада протеолитических реакций, приводящих к расщеплению белков ядерного матрикса, дестабилизации структуры хроматина ядер, фрагментации ДНК, нарушению репликационной и метаболической функций клетки. Среди ферментов, причастных к разветвленной цепи апоптоза, ведущее место принадлежит каспазам, относящимся к семейству 1-интерлейкин-конвертирующих протеаз.
5. Участие специфических регуляторных белков, препятствующих реализации апоптозной программы. Один из них - белок bcl-2 оказывает защитное действие на многие типы клеток, пребывающие в неблагоприятных условиях. В исследованиях in vitro для оценки апоптоза используется отношение белков bcl-2/Bax; избыток первого способствует выживанию клетки, избыток второго - ее гибели.
Баланс про- и антиапоптических механизмов связан с оксидом азота (NO), различные пути образования которого могут определять как цитотоксическое, так и цитопротекторное его действие. Длительная генерация NO провоцирует развитие апоптоза: усиливается синтез белка р53 и активируются протеазы из семейства каспаз.
Принято считать, что каскад апоптозных процессов может быть спровоцирован либо прямым действием на геном клетки (вирусы), либо через нейромедиаторы (глутамат), либо причинами, связанными с ишемией клетки, ее физическим повреждением, реперфузией, токсическим воздействием. Биохимические процессы, сопровождающие апоптоз, проявляются экспрессией специфических генов и синтезом особых белков клетки, запускающих реакции апоптоза. Число таких "инициаторов", связанных с патологиями различной этиологии, оказывается значительным. Такая полиэтиологичность предопределяет инициацию апоптоза при многих патологических состояниях - как всего организма, так и отдельных его органов или клеточных популяций (см. рис.).
Биохимические и цитоморфологические исследования апоптоза выявляют несколько стадий его развития в поврежденной нервной ткани: непосредственную и отсроченную. Как правило, исходное повреждение ткани не ограничивается областью воздействия разрушающей силы, а, продолжаясь во времени, захватывает первично интактные клетки и приводит к расширению очага повреждения.
Поскольку апоптоз рассматривается в качестве патохимического механизма клеточной гибели, представляет собой фазный процесс и, следовательно, имеет обратимые этапы, возможно рассмотрение подходов к фармакологическому вмешательству в его регуляцию. Существенной оказывается оценка условий, благодаря которым, переходя необратимую грань, апоптоз приводит к тотальным последствиям, гибели большого массива функционально значимых клеток и смерти всего организма. К этим условиям относятся характер и величина травмирующего воздействия, степень сбалансированности клеточных биохимических систем, противостоящих необратимому развитию апоптоза, возможность своевременного применения реабилитационных мероприятий защиты структур мозга, кардиоваскулярной системы и организма в целом.
РОЛЬ НЕЙРОПЕПТИДОВ И НЕЙРОТРОФИНОВ
Существует определенная параллель между информацией об участии нейропептидов и нейротрофических факторов роста в патогенезе неврологических расстройств и причастностью этих химических регуляторов к нейроапоптозу. Среди нейротрофических ростовых факторов (см. "МГ" № 30 от 17.04.02) выявляются те, которые играют роль индукторов апоптических процессов или, наоборот, противодействующих развитию нейроапоптоза. Например, в исследованиях последних месяцев подтверждено участие фактора некроза опухоли (TNF-() в апоптозе глиальных и нейрональных клеток как следствии аутоиммунной нейропатологии и патологии мультисклероза. Ассоциируемое с болезнью Гентингтона образование свободнорадикальных продуктов, провоцируемое в эксперименте внесением дофамина, ингибировалось в нейронах стриатума нейротрофическим фактором мозга (BDNF).
Новым направлением в исследовании нейропептидов стало определение их роли в регуляции апоптоза. Наряду с данными, свидетельствующими об участии вазоактивного пептида эндотелина-1 и его рецепторов (ЕТА) в ишемической патологии мозга, получена информация об антиапоптической активности этого пептида. На ряде моделей нейроапоптоза было также продемонстрировано защитное действие кальцитонинового нейропептида (CGRP) и пептидного фрагмента ангиотензина IV. В то же время было установлено, что сам ангиотензин II, также как и пептид кальцийнейрин, напротив, способствует индукции проапоптического каскада. Эти факты, демонстрирующие значимость нейропептидов и ростовых факторов в нормальной и патологической деятельности мозга, отражают организацию поливариантной системы химической регуляции, обеспечивающей как жизнеспособность и защиту нейронов от неблагоприятных влияний, так и программируемую гибель определенной части клеточной популяции в случае повреждения мозга. Открытие нейротрофических пептидных факторов побудило к формированию новой стратегии - пептидергической, или нейротрофной терапии нейродегенеративных патологий. Исходная идеология связывает нейродегенеративные патологии, включая болезнь Альцгеймера, с активностью различных нейротрофических факторов мозга и нейропептидов. На этой основе был разработан ряд препаратов, успешно применяемых в терапии большого спектра неврологических расстройств. Наибольший успех здесь выпал на долю церебролизина, уже в течение двух десятков лет успешно используемого в клинике неврологических и психиатрических заболеваний.
АПОПТОЗ В КАРДИОВАСКУЛЯРНЫХ КЛЕТКАХ
Признаки апоптоза выявляются также при различных формах сердечно-сосудистых патологий: дисфункции эндотелия, "чрезмерном" напряжении сосудистой стенки, ишемических и реперфузионных ее нарушениях, атеросклеротических изменениях, инфаркте миокарда, ишемическом инсульте и др. В перикардиальной жидкости пациентов с ишемической патологией миокарда обнаружен митогенактивирующий белок р38, участвующий в запуске апоптических процессов. Известно также, что при кардиоваскулярных расстройствах, сочетающихся с диабетом, в миоцитах и эндотелиоцитах обнаруживаются активированные апоптозные продукты. Недавно были опубликованы клинические результаты, согласно которым определенные белки апоптозного каскада могут быть использованы как маркеры повреждения миокарда при стадийной ИБС.
Физиологически активные пептиды (ангиотензин II, эндотелин-1, брадикинин, адреномедуллин и натрийуретические факторы) вносят свой "вклад" в развитие процессов, связанных с дисфункцией эндотелия и развитием клеточного апоптоза. Очевидно, немаловажная роль принадлежит также нейротрофическим ростовым полипептидам (фактору опухолевого роста, трансформирующему ростовому фактору, фактору роста фибробластов, эндотелиальному фактору роста сосудов, инсулиноподобному фактору роста и др.), а также специфическим белкам клеточной адгезии и мембранного взаимодействия - интегринам и селектинам. Регуляторные пептиды и нейротрофические ростовые факторы могут выполнять функцию как проапоптических (провоцирующих, индуцирующих), так и антиапоптических агентов при кардиоваскулярной патологии. Их соотношение играет ключевую роль в стадийном формировании необратимых повреждений миоцитов, эндотелиальных и васкулярных клеток и превращения патологии в глобальный феноптический процесс.
Как известно, патология "гипертонического сердца" сопровождается гипертрофией левого желудочка, ведущей к ремоделированию миокарда, недостаточной васкуляризации, фиброзу, уменьшению числа функциональных кардиомиоцитов и сократительной способности сердца в целом. За все эти явления ответствены апоптозные процессы, спровоцированные хронической ишемией и перегрузкой больного сердца. Исследования последнего периода показывают, что апоптозная гибель кардиомиоцитов служит решающим фактором в переходе к компенсаторной гипертрофии и нарушению насосной функции сердца при артериальной гипертензии. Условия, провоцирующие экспрессию химических механизмов апоптоза в "перегруженном" сердце, включают чрезмерное механическое напряжение миоцитов, гипоксию и окислительный стресс, активацию нейрогуморальных факторов и цитокинов. Все эти явления сопряжены между собой и составляют звенья единого патогенетического процесса.
Особенно значимы исследования, связанные с ангиотензином II и образующим его ферментом АПФ. Исследования роли ангиотензина II в механизмах окислительного стресса и эндотелиальной дисфункции логично привели к рассмотрению роли этого пептида в индукции апоптоза кардиальных, васкулярных и эндотелиальных клеток. Принципиальным оказывается вывод, что повышенный уровень ангиотензина II сопряжен с активностью некротического и трансформирующего ростовых факторов, а также специфических апоптозных белков (bcl-2, p53), особых ферментов (каспаз), каскадная динамика которых приводит к деградации молекулы ДНК на фрагменты и последующей "запрограммированной" гибели клетки.
Проапоптозный, патогенетический, эффект ангиотензина II может быть заблокирован использованием соответствующих препаратов.
Рассматривая проблему апоптоза как сложный биологический и патохимический феномен, можно определить, что как нейропептиды, так и нейротрофические факторы представляют в данном случае показательный пример участия в поливариантной системе контроля функций на всех "этажах" физиолого-биохимической организации. Эти химические соединения служат элементами глобальной информационно-регуляторной сети организма, определяющей его жизнь и смерть.
В настоящее время открыт, по сути, базовый биохимический механизм неврологических, кардиоваскулярных, онкологических и других патологий. Очевидно, последующие работы и исследования специфической роли систем химических регуляторов представят новые возможности диагностики и терапии этих заболеваний.

Апоптоз - программированная клеточная гибель, энергетически зависимый, генетически контролируемый процесс, который запускается специфическими сигналами и избавляет организм от ослабленных, ненужных или повреждённых клеток. Ежедневно, примерно около 5% клеток организма подвергаются апоптозу, а их место занимают новые клетки. В процессе апоптоза клетка исчезает бесследно в течение 15-120 минут.
Запрограммированная клеточная гибель это биохимически специфический тип гибели клетки, который характеризуется активацией нелизосомных эндогенных эндонуклеаз, которые расщепляют ядерную ДНК на маленькие фрагменты. Морфологически апоптоз проявляется гибелью единичных, беспорядочно расположенных клеток, что сопровождается формированием округлых, окруженных мембраной телец (“апоптотические тельца”), которые тут же фагоцитируются окружающими клетками.
Апоптоз – энергозависимый процесс, посредством которого удаляются нежелательные и дефектные клетки организма. Он играет большую роль в морфогенезе и является механизмом постоянного контроля размеров органов. При снижении апоптоза происходит накопление клеток, пример – опухолевый рост. При увеличении апоптоза наблюдается прогрессивное уменьшение количества клеток в ткани, пример – атрофия.
Морфологические проявления апоптоза.
Апоптоз имеет свои отличительные морфологические признаки, как на светооптическом, так и на ультраструктурном уровне. При окраске гематоксилином и эозином апоптоз определяется в единичных клетках или небольших группах клеток. Апоптотические клетки выглядят как округлые или овальные скопления интенсивно эозинофильной цитоплазмы с плотными фрагментами ядерного хроматина. Поскольку сжатие клетки и формирование апоптотических телец происходит быстро и также быстро они фагоцитируются, распадаются или выбрасываются в просвет органа, то на гистологических препаратах он обнаруживается в случаях его значительной выраженности. К тому же апоптоз – в отличие от некроза – никогда не сопровождается воспалительной реакцией, что также затрудняет его гистологическое выявление.
Наиболее четко морфологические признаки выявляются при электронной микроскопии. Для клетки, подвергающейся апоптозу характерно:
Сжатие клетки. Клетка уменьшается в размерах; цитоплазма уплотняется; органеллы, которые выглядят относительно нормальными, располагаются более компактно. Предполагается, что нарушение формы и объема клетки происходит в результате активации в апоптотических клетках трансглютаминазы. Этот фермент вызывает прогрессивное образование перекрестных связей в цитоплазматических белках, что приводит к формированию своеобразной оболочки под клеточной мембраной, подобно ороговевающим клеткам эпителия.
Конденсация хроматина. Это наиболее характерное проявление апоптоза. Хроматин конденсируется по периферии, под мембраной ядра, при этом образуются четко очерченные плотные массы различной формы и размеров. Ядро же может разрываться на два или несколько фрагментов. Механизм конденсации хроматина изучен достаточно хорошо. Он обусловлен расщеплением ядерной ДНК в местах, связывающих отдельные нуклеосомы, что приводит к развитию большого количества фрагментов, в которых число пар оснований делится на 180-200. При электрофорезе фрагменты дают характерную картину лестницы. Эта картина отличается от таковой при некрозе клеток, где длина фрагментов ДНК варьирует.
Формирование в цитоплазме полостей и апоптотических телец. В апоптотической клетке первоначально формируются глубокие впячивания поверхности с образованием полостей, что приводит к фрагментации клетки и формированию окруженных мембраной апоптотических телец, состоящих из цитоплазмы и плотно расположенных органелл, с или без фрагментов ядра.
Фагоцитоз апоптотических телец. Фагоцитоз апоптотических клеток или телец осуществляется окружающими здоровыми клетками, или паренхиматозными, или макрофагами. Апоптотические тельца быстро разрушаются в лизосомах, а окружающие клетки либо мигрируют, либо делятся, чтобы заполнить освободившееся после гибели клетки пространство. Фагоцитоз апоптотических телец макрофагами или другими клетками активируется рецепторами на этих клетках: они захватывают и поглощают апоптотические клетки. Один из таких рецепторов на макрофагах – рецептор витронектина, который является β3-интегрином и активирует фагоцитоз апоптотических нейтрофилов.
Участие апоптоза в физиологических и патологических процессах
Запрограммированном разрушении клеток во время эмбриогенеза (включая имплантацию, органогенез). Несмотря на то, что при эмбриогенезе апоптоз не всегда является отражением “запрограммированной смерти клетки”, это определение апоптоза широко используют различные исследователи.
Гормон-зависимой инволюции органов у взрослых, например, отторжение эндометрия во время менструального цикла, атрезии фолликулов в яичниках в менопаузе и регрессия молочной железы после прекращения лактации.
Удалении некоторых клеток при пролиферации клеточной популяции.
Гибели отдельных клеток в опухолях, в основном при ее регрессии, но также и в активно растущей опухоли.
Гибели клеток иммунной системы, как В -, так и Т-лимфоцитов, после истощения запасов цитокинов, а также гибели аутореактивных Т-клеток при развитии в тимусе.
Патологической атрофии гормон-зависимых органов, например, атрофии предстательной железы после кастрации и истощении лимфоцитов в тимусе при терапии глюкокортикоидами.
Патологической атрофии паренхиматозных органов после обтурации выводных протоков, что наблюдается в поджелудочной и слюнных железах, почках.
Гибели клеток, вызванных действием цитотоксических Т-клеток, например, при отторжении трансплантата и болезни “трансплантат против хозяина”.
Повреждении клеток при некоторых вирусных заболеваниях, например, при вирусном гепатите, когда фрагменты апоптотических клеток обнаруживаются в печени, как тельца Каунсильмана.
Гибели клеток при действии различных повреждающих факторов, которые способны вызвать некроз, но действующих в небольших дозах, например, при действии высокой температуры, ионизирующего излучения, противоопухолевых препаратов.
Биохимия апоптоза.
Кроме того, в апоптозе принимают участие и другие протеазы, прежде всего, кальпаины, или Са2+-зависимые протеазы и убиквитин (протеаза, ковалентно связывающаяся с белком-мишенью). Эти протеазы — обязательный компонент каскада протеолитических ферментов. Так, ингибиторы кальпаина блокируют апоптоз. Убиквитин-протеосомный путь деградации белков активируется при апоптозе.
Роль каспаз в апоптозе разнообразна. Результатом активности протеаз являются характерные изменения в морфологии клеток при апоптозе.1. Гидролиз белков ламинов, армирующих ядерную мембрану. Это ведет к распаду ядерной оболочки и конденсации хроматина. Мишенями протеаз при апоптозе являются также белки ядрышек, гистоны и негистоновые белки и топоизомераза. Топоизомераза — связующее звено между ДНК хроматина и белковыми структурами ядра, с помощью которого хроматин прикрепляется к ядерному матриксу. Расщепление топоизомеразы — это этап образования высокомолекулярных фрагментов ДНК.
2. Расщепление антиапоптозных белков — протеолиз ингибитора ДНКазы, ответственной за фрагментацию ДНК. В нормальных клетках апоптозная ДНКаза CAD (caspase-activated DNase) образует неактивный комплекс с ингибитором 1CMiwm DFF (DNA fragmentation factor). При апоптозе ингибитор Гмс участием каспаз 3 и 7 инактивируется и свободная CAD, вызывая нуклеосомные разрывы хроматина, ведет к образованию фрагментов ДНК с молекулярной массой кратной молекулярной массе ДНК в нуклеосомных частицах — 180-200 пар нуклеотидов. Эти фрагменты и дают характерную лесенку ДНК при электрофоретическом разделении в агарозном геле. Апоптоз возможен и без фрагментации ДНК. Обнаружен ядерный белок ACCINVS (apoptotic chromatin condensation inducer in the nucleus), который при комбинированном действии каспазы 3 и неидентифицированной протеазы расщепляется на фрагменты. Один из них в присутствии дополнительных неядерных факторов вызывает апоптотическую конденсацию хроматина и фрагментацию ядра (кариорексис) без фрагментации ДНК. Кроме непосредственной активации нуклеаз, протеазы (путем ограниченного протеолиза) устраняют структурное разобщение между нуклеазами и ДНК в составе хроматина, удаляют белки, защищающие ДНК.3. Угнетение репарации ДНК: инактивирование и нарушение регуляции белка, участвующего в репарации ДНК, а также в сплайсинге мРНК, репликации ДНК. Мишенью каспаз является поли-(АДФ-рибозо)-полимераза (ПАРП), которая участвует в репарации ДНК (катализирует полиАДФ-рибозилирование белков, связанных с ДНК). Донором АДФ-рибозы является NAD'. Активность ПАРП-полимеразы возрастает в 500 раз и более при связывании с участками разрыва ДНК. ПАРП участвует в репарации поврежденной ДНК, регуляции активности эндонуклеаз, поддержании структуры хроматина посредством АДФ-рибозилирования. Апоптотическая гибель клетки сопровождается расщеплением ПАРП каспазами. При массированных разрывах ДНК чрезмерная активация ПАРП, сильно снижая содержание внутриклеточного NAD*, ведет к подавлению гликолиза и митохондриального дыхания и вызывает гибель клетки по пути некроза.4. Разрушение белков цитоскелета. Деградация структурных и функциональных белков митотического аппарата.5. Участие в экспрессии генов. Эта функция связана с протеолизом репрессоров и с образованием пептидов, регулирующих транскрипцию (модификация факторов транскрипции). Субстратом протеаз является, например, гистон, выступающий репрессором генов.6. Одна из функций протеаз — передача апоптозного сигнала от индукторов апоптоза. Сигналы могут быть трансмембранными, рецептор-зависимыми. Рецепторами служат трансмембранные белки. Протеазы принимают участие либо непосредственно при взаимодействии индукторов апоптоза с рецепторами, либо через активацию протеинкиназ, играющих важную роль в передаче трансмембранного сигнала с целого ряда рецепторов.Локализация протеаз в различных отделах (компартментах) клетки способствует эффективной трансмембранной передаче сигналов программируемой клеточной гибели. Часть протеаз связаны с мембранами (цитоплазматической, ядерной, мембранами органелл или вакуоли) — это мембраносвязанные протеазы. Другие — находятся в матриксе ядра, цитоплазмы или органелл. Аспарагиновая протеаза растений, по всей видимости, локализована в вакуоли. Предполагается, что сериновые протеазы локализуются в цитоплазме и в ядре. Известно, что в ядрах протеазы могут быть прочно ассоциированы с хроматином и, в том числе, непосредственнно с гистонами. Перемещение протеаз в клетке может сопровождаться их активацией. Например, повышение концентрации Ca2+ внутри клетки способствует перемещению Са2+-зависимой протеазы и протеинкиназы из цитоплазмы в мембрану. При этом происходит автокаталитическая активация неактивных форм протеазы.Так, активация некоторых протеаз может быть обусловлена увеличением концентрации кальция в клетках, наблюдаемой при разных типах апоптоза (раздел выше). АФК также могут быть непосредственными индукторами активации протеаз. Появление локальных участков однонитевой ДНК активирует, например, ядерные ДНК-зависимые сериновые протеазы, специфичные к гистону.
Множество ветвей сигнальной трансдукции перепроверяет правильность выбранного алгоритма событий на пути к апоптозу, уберегая клетку от бессмысленной гибели. Выявлено несколько механизмов, ограждающих клетку от случайного самоуничтожения с участием протеаз.
Во-первых, протеазы синтезируются в клетке в неактивной форме, а процессинг неактивных форм протеаз происходит путем автолиза или путем протеолиза другими протеазами. Например, каспазы синтезируются в клетке в виде прокаспазы — неактивного мономера с молекулярной массой 30-50 кДа. Активные формы — тетрамеры, содержащие по две субъединицы: (р 10 — р20)2 (рис. 9.7). Прокаспазы обладают незначительной протеолитической активностью, составляющей 1-2% активности зрелой каспазы. Механизм протеолитического само- или перекрестного расщепления (ауто- или транс-процессинга), а затем пространственного сближения ведет к образованию активных каспаз. От прокаспазы отделяется регуляторный N-концевой домен (продомен), а оставшаяся часть молекулы разделяется на большую (около 20 кДа) и малую (около 10 кДа) субъединицы. Затем происходит ассоциация большой и малой субъединиц. Два гетеродимера образуют тетрамер с двумя каталитическими центрами, работающими независимо. Первоначально концентрация каспаз в клетке ничтожна. Благодаря свойству автокатализа, концентрация активных каспаз может возрастать лавинообразно.Во-вторых, протеазы обратимо взаимодействуют с эндогенными белковыми ингибиторами, образуя неактивные комплексы (латентные комплексы описаны для цистеиновых, Са2+-зависимых и некоторых других протеаз). При действии различных индукторов апоптоза происходит диссоциация неактивных комплексов протеаза-ингибитор. Обратимое взаимодействие Са2+-зависимых протеаз с эндогенными ингибиторами регулируется кальцием. Цистеиновая протеаза связывается ковалентно с ингибитором через дисульфидную связь. Высвобождение и активация каспазы происходит в результате тиол-дисульфидного обмена и сопряжена с окислительно-восстановительным состоянием клетки и метаболизмом глюкозы.В-третьих, протеазы могут быть компонентами специальных рецептор-зависимых систем. Так, [рецептор + лиганд + адаптер + прокаспаза] формируют специфический агрегат, в котором происходит активация каспаз. Такой агрегат называют апоптосомой или апоптозным шапероном. Самое интересное, что выявлены консервативные области гомологии (в том числе NB-область) белка адаптера у животных и продуктов генов резистентности у растений, включая томат, арабидопсис и табак. Более того, белки похожи структурно. Предполагается, что продукты гена резистентности могут играть роль адаптеров в апоптосоме. Таким образом, при узнавании продукта авирулентности, по всей видимости, происходит диссоциация апоптосомы и развертывание программы апоптоза.
Продукты генов резистентности, по-видимому, ответственны за эффективность гибели клеток при заражении — узнавание факторов и запуск машины самоуничтожения, за первые (самые важные) шаги на пути к стремительной гибели клетки.Существует несколько путей реализации программы ПКГ. Путь передачи сигнала: индукторы — рецепторы — адаптеры — каспазы первого эшелона — регуляторы — каспазы второго эшелона. Рецептор взаимодействует с лигандом. Насколько обратима гибель клетки? На этапе активации каспаз первого эшелона жизнь клетки еще можно сохранить. Существуют регуляторы, которые блокируют или, напротив, усиливают разрушительное действие каспаз первого эшелона. После активации каспазами первого эшелона каспаз второго эшелона путем протеолиза из прокаспаз процесс, запушенный программой смерти, становится необратим. Эти каспазы способны в дальнейшем к самоактивации (автокатализу или автопроцессингу) и активируют фактор фрагментации ДНК на нуклеосомные фрагменты. Вернемся к митохондриям. Апоптотическое изменение митохондрии может индуцироваться окислительным стрессом, повышением концентрации Ca2+. При апоптозе из межмембранного пространства митохондрий высвобождаются белки — апоптогенные факторы:
AIF (Apoptosis Inducing Factor) — флавопротеин с молекулярной массой 57 кДа. Будучи добавлен к изолированным ядрам, он вызывает конденсацию хроматина и фрагментацию ДНК, а при добавлении к изолированным митохондриям — высвобождение цитохрома С и каспазы 9. Высвобождаемый цитохром С вместе с цитоплазматическим фактором APAF-1 (apoptosis protease activating factor-1) образует комплекс с прокаспазой. APAF-I играет роль арматуры, на которой происходит аугокаталитический процессинг каспазы 9 (мультимерная арматура APAF1-цитохром-С-комплексов напоминает пропеллер). Обнаружены ингибиторы высвобождения цитохрома С, блокирующие апоптоз, например, белок Bel.
Список используемой литературы:
Гордеева А.В., Лабас Ю.А., Звягильская Р.А.Апоптоз одноклеточных организмов: механизмы и эволюция Биохимия, 2004, том 69, вып. 10, с. 1301—1313
Голубев А.М., Москалева Е. Ю., Северин С.Е., Веснянко Т.П., Кузовлев А.Н., Алкадарский А.С., Порошенко Г.Г. Апоптоз при критических состояниях

Выживание многоклеточных организмов требует состояния равновесия между пролиферацией и отмиранием клеток. Аномальная регуляция апоптоза (АП) подразумевается при возникновении и развитии практически всех заболеваний. Многие расстройства в процессе своего патогенеза можно классифицировать по признаку их взаимосвязи с избыточным или недостаточным АП клеток разного вида. В работе обсуждается значение нарушений АП в патогенезе ряда заболеваний.
Все аутоиммунные заболевания можно рассматривать в качестве принципиальных недостатков АП, так как отмирание аутореактивных лимфоцитов зависит от этого процесса. Исследования в области изучения функций молекул Fas и FasL позволяют сформировать новый взгляд на патогенез и лечение аутоиммунного сахарного диабета. В процессе его развития основным недостатком процесса регулирования АП является отсутствие аутотолерантности, что приводит к недостаточному апоптозному уничтожению аутореактивных Т-клеток. В присутствии опасных лимфоцитов, клетки поджелудочной железы разрушаются. Разрегулирование процесса выражения Fas и FasL, сопровождающее инфекции, может привести к тому, что клетки тканей будут индуцировать у своих соседей АП в форме локального аутоиммунитета. Патогенез тиреоидита Хашимото основан на системе Fas/FasL. Стимулирование тироцитов цитокинами при инфекции или иной форме воспаления приводит к выражению Fas вместе с FasL на тироцитах. Такая неблагоприятная ситуация может привести к уничтожению тироцитов друг другом, объясняя высокий уровень распространённости АП среди случаев тиреоидита Хашимото. При гепатитах стимулируемый Fas процесс отмирания гепатоцитов может быть блокирован специальным фармакологическим механизмом подавления каспаз.
Отсутствие аутотолерантности Т-лимфоцитов - в связи с недостаточным развитием либо анергии, либо стимулированного Fas АП - также может стать основой патогенеза лимфопролиферативных расстройств. Если аутореактивные лимфоциты возбуждаются и своевременно не удаляются или не становятся толерантными, то могут увеличить вероятность аутоиммунной реакции. Генные мутации Fas/FasL сопровождаются аутоиммунными расстройствами и повышенным уровнем лимфоцитов. Негативные и рецессивные мутации в гене Fas, которые препятствуют трансдукции апоптозного сигнала, обнаружены у пациентов с аутоиммунными лимфопролиферативными синдромами. У таких больных выявлены: скопление доброкачественных недоразвитых Т-лимфоцитов во вторичных лимфоидных органах (с увеличением до заметной аденопатии), поликлональная гипергаммаглобулинемия, выработка аутоантител, тромбоцитопения, нейтропения и гломерулонефрит.
АП имеет большое значение, как для патогенеза многих раковых болезней, так и резистентности их к противоопухолевым методам лечения. Мутации в гене p53 и его регуляторах являются широко распространёнными у онкологических больных. Подробное исследование АП открывает новую страницу в понимании и лечении рака. Отмирание апоптозной клетки играет роль во многих нейродегенеративных расстройствах, включая болезни Альцгеймера и Паркинсона, мышечную атрофию спинного мозга, боковой амиотрофический склероз. Проблема развития АП и уровней его молекулярных медиаторов при кардиологических болезнях человека становится всё более актуальной, так как уровень АП в больных сердцах высокий. Понимание молекулярных механизмов АП представляет собой основу предпосылок для развития современных методов лечения.
Апоптоз представляет собой систему контроля клеточной дифференцировки, обеспечивающую самоуничтожение дефектных структур. Тем самым поддерживая нормальное функционирование целостного организма. Регуляторами апоптоза являются белки семейства Bcl-2, которые могут обладать как антиапоптическими свойствами, так и проапоптическими. Проапоптические белки запускают синтез цитохрома С в миохондриях, провоцирующего образование апоптосомы, активирующей каспазы, которые обладают способностью к денатурации белков, что приводит к гибели клетки. Для обнаружения дефектов в клетке существует белок р53, который при наличии повреждений в клетке активирует синтез проапоптических белков. При недостаточном апоптозе, дефектные клетки пролиферируют, что приводит к их злокачественной трансформации. Злокачественные клетки подвергаются серии генетических изменений. Если это способствует их преимущественному росту над нормальными клетками, то риск развития новообразований значительно возрастает. Потеря предшественника апоптоза - белка P53 является наиболее распространенным механизмом уклонения от гибели. Канцерогенез можно рассматривать как сложный клеточный процесс, который связан с неограниченным репликативным потенциалом, независимостью от сигналов роста и параллельным сопротивлением ингибирующему рост сигналу, уклонение от активации клеточной смерти, устойчивый ангиогенез, а также способность тканевой инвазии и метастазирования.

1. Кузнецов, С.Л. Гистология, цитология и эмбриология / С.Л. Кузнецов, Н.Н. Мушкамбаров Н. – М.: МИА, 2007.
2. Галицкий В.А. Возникновение эукариотических клеток и происхождение апоптоза // Цитология, 2008, том 47, вып. 2, с. 103–120
3. Hanahan D, Weinberg RA: The hallmarks of cancer. Cell. 2000, 100: 57-70. 10.1016/S0092-8674(00)81683-9.
4. Taylor, R. C., Cullen, S. P., and Martin, S. J. (2008). Apoptosis: controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 9, 231–241.
5. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 2014; 15: 49–63.
7. Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015.
8. McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol 2013; 5: a008656.
9. Manzl C, Fava LL, Krumschnabel G, Peintner L, Tanzer MC, Soratroi C et al. Death of p53-defective cells triggered by forced mitotic entry in the presence of DNA damage is not uniquely dependent on Caspase-2 or the PIDDosome. Cell Death Dis 2013; 4: e942.
10. Shalini S, Dorstyn L, Dawar S and Kumar S: Old, new and emerging functions of caspases. Cell Death Differ. 22:526–539. 2015.
Клеточный гомеостаз в организме здорового человека определяется балансом между гибелью и пролиферацией клеток. Дефекты, возникающие в процессах дефференцировки и новообразования клеток, ведут к самоуничтожению этих структур [2]. Может показаться парадоксальным, что стимуляция клеточной гибели может способствовать выживанию организма.
Механизм, отвечающий за инициирование и выполнение запрограммированной гибели клеток, называется апоптозом. Он осуществляется под действием внеклеточных или внутриклеточных факторов. Под воздействием этого процесса, ДНК распадается на фрагменты, клетка сжимается, клеточные мембраны разрушаются, происходит элиминация и она поглощается соседней клеткой или специфичной клеткой имунной системы. Особенностью этого процесса является то, что мембрана клетки не разрушается до полного завершения этапов самопроизвольной гибели. Что дает возможность избежать риска возникновения воспалительных процессов. Обычно от начала запуска апоптоза до окончательной клеточной фрагментации требуется несколько часов. Однако этот период зависит от типа клетки, стимула и апоптотического пути [1,2].
Апоптозные клетки выглядят как округлые либо овальные скопления интенсивно эозинофильной цитоплазмы с плотными фрагментами ядерного хроматина [1]. (Рис. 1.).
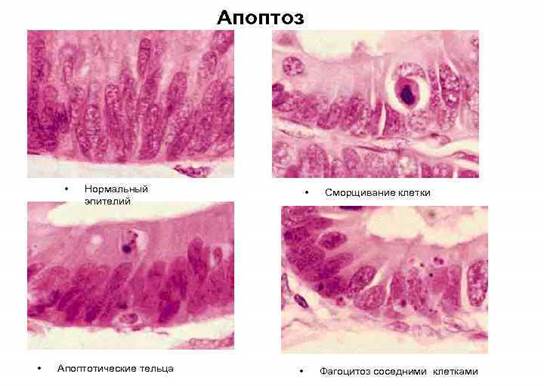
Рис. 1. Стадии апоптоза эпителиальной клетки
Основными регуляторами запрограммированной гибели клеток являются белки, принадлежащие к семейству Bcl-2. Эти белки могут, как активировать апоптоз, то есть быть проапоптотическими, так и ингибировать его, обладая антиапоптотическими свойствами. Антиапоптотические белки в здоровой клетке связывают и инактивируют проапоптотические. Это происходит тогда, когда она не нуждается в гибели [4,5].
Контроль такой элиминации производится в митохондриях, которые обеспечивают внутреннюю часть апоптического пути. В этих органеллах содержатся сигнальные молекулы, связаные с митохондриальной мембраной и известные как цитохром C. В ответ на проапоптотические сигналы из проапоптотических белков, высокую концентрацию Ca 2+ в цитозоле или гипоксию цитохром C высвобождается в клетку митохондриями и связывается с белком. Это приводит к образованию апоптосомы. После образования апоптосома активирует группу белков под названием каспазы, которые денатурируют другие белки в клетках [8]. Так как активные каспазы могут разрушающе воздействовать внутри здоровой клетки, они производятся в неактивной форме – прокаспазы. В фазе апоптоза семейство каспаз представляет собой основные эффекторные молекулы самого процесса элиминации, которые вносят вклад в конечные стадии апоптотической гибели клеток путем компонентов цитоскелетного аппарата и ядерной ДНК [4,9]. Все каспазы подразделяются на инициаторы, эффекторы и стимуляторы. Инициаторы расщепляют и активируют каспазы эффекторы, амплифицируя сигнал. Эффекторы расщепляют различные белки, что приводит к процессу апоптоза. Активация каспаз ведет к запуску протеолитического каскада реакций, провоцирующих гибель клетки [9].
Помимо каспаз, чрезвычайно важным является белок P53, обеспечивая обнаружение повреждения ДНК, аномалий хромосом и остановку клеточного цикла. Если повреждения необратимые, то апоптоз актимируется. P53 активирует процесс путем увеличения продуцирования проапоптотического белка, который активирует каспазный каскад, что в конечном итоге приводит к самоуничтожению клетки [10].
Апоптоз — это защита организма от персистенции пораженных клеток, которые могут оказаться потенциально опасными для многоклеточного организма. Однако, эти процессы могут нарушаться, сопровождаясь либо чрезмерным апоптозом, например, в случае дегенеративных заболеваний, либо недостаточным, что приводит к ускоренной пролиферации дефектных клеток и возникновению онкологии [2]. Проблемы недостаточного самоуничтожения клеток могут возникать на любом этапе апоптоза, что приводит к злокачественной трансформации пораженных клеток, метастазированию опухолей и устойчивости к противоопухолевым препаратам [6]. Следовательно, устойчивость к апоптозу или угнетение этого процесса играют жизненно важную роль в канцерогенезе. Одной из причин торможения процессов самопроизвольной гибели клеток является нарушения баланса проапоптотических и антиапоптотических белков. Проапоптотические (например, Bax, Bad, Bcl-Xs) и антиапоптотические белки (например, Bcl-2, Bcl-XL, Mcl-L и т. Д.) являются двумя основными группами белков семейства Bcl -2. Антиапоптотические белки регулируют апоптоз, блокируя выделение цитохрома С в митохондриях, в то время как проапоптические -стимулируют . Нарушение этого баланса вызывает угнетение процесса апоптоза в пораженных клетках. Причиной может стать как чрезмерная экспрессия антиапоптотических белков, так и проапоптотических или их комбинации.
Так, в основе возникновения В-клеточной лимфомы лежит механизм, подавляющий синтез проапоптического белка семейства Bcl-2, что приводит к торможению апоптоза клеток фолликулярной лимфомы и их пролиферацию [3]. (Рис. 2.).
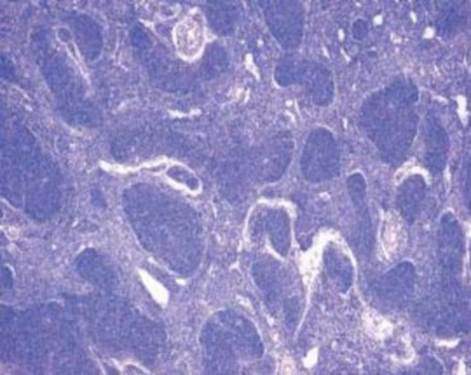
Рис. 2. Гистопрепарат ткани лимфатического узла. Фолликулярная лимфома.
Новообразования можно рассматривать как результат последовательности генетических изменений, в течение которых нормальная клетка превращается в злокачественную. И именно несостоявшийся апоптоз таких клеток является одним из существенных критериев, которые вызывают злокачественную трансформацию [3]. Злокачественные клетки подвергаются серии генетических изменений. Если это способствует их преимущественному росту над нормальными клетками, то риск развития и роста новообразований значительно возрастает. Например, когда в клетках кожи возникают повреждения под воздействием ультрафиолетового излучения (например, солнцем, соляриями), обычно срабатывает апоптоз. Это помогает устранить патологические элементы. Если апоптоз не происходит, такие клетки могут выживать и пролифирировать, превращаясь в злокачественные. (Рис.3.).
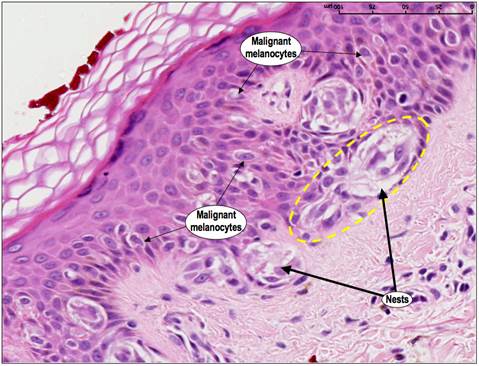
Выводы. Таким образом, канцерогенез можно рассматривать как сложный клеточный процесс, который связан с неограниченным репликативным потенциалом, независимостью от сигналов роста и параллельным сопротивлением ингибирующему рост сигналу, уклонение от активации клеточной смерти, устойчивый ангиогенез, а также способность тканевой инвазии и метастазирования. Злокачественные опухоли являются инвазивными и могут метастазировать в отдаленные места через систему кровообращения. Следовательно, метастатическое распространение, а не первичная опухолевая нагрузка, является основной причиной смертей от рака [6,4,7].
Читайте также: