
Ультрафиолетовая электронная спектроскопия реферат
Обновлено: 08.05.2024
Реферат на тему: УФ - спектроскопия.
Качественный анализ и идентификация веществВыполнила:
Студентка 2 курса
Фармацевтического факультета6 группы
Арушанян Армине
Преподаватель: Варинский Б.А.
Запорожье 2013Содержание:
1) УФ – спектроскопия.
2) Качественный анализ.
3) Идентификация веществ.
Ультрафиолетовая спектроскопия
УЛЬТРАФИОЛЕТОВАЯ СПЕКТРОСКОПИЯ (УФ спектроскопия, УФС), раздел оптич. спектроскопии, включающий получение, исследование иприменение спектров испускания, поглощения и отражения в ультрафиолетовой области, т. е. в диапазоне длин волн 10-400 нм (волновых чисел 2,5 · 104 - 106 см-1). УФС при длине волны меньше 185 нм наз. вакуумной, т. к. в этой области УФ излучение настолько сильно поглощается воздухом (гл. обр. кислородом), что необходимо применять вакуумные или наполненные непоглощающим газом спектральные приборы.
Техникаизмерения УФ спектров в осн. такая же, как спектров в видимой области (см. Спектрофотометрия). Спектральные приборы для УФС отличаются тем, что вместо стеклянных оптич. деталей применяют аналогичные кварцевые (реже флюоритовые или сапфировые), к-рые не поглощают УФ излучение. Для отражения УФ излучения используют алюминиевые покрытия. Приемниками служат обычные или маложелатиновые фотоматериалы, атакже фотоэлектрич. приборы, гл. обр. фотоэлектронные умножители, счетчики фотонов, фотодиоды, ионизационные камеры. Для увеличения чувствительности при использовании фотоматериалов иногда регистрируют флуоресценцию, вызванную исследуемым УФ излучением.
Качественный анализ имеет своей целью обнаружение определенных веществ или ихкомпонентов в анализируемом объекте. Обнаружение проводится путем идентификации веществ, то есть установления тождественности (одинаковости) АС анализируемого объекта и известных АС определяемых веществ в условиях применяемого метода анализа. Для этого данным методом предварительно исследуют эталонные вещества (гл. 2.1), в которых наличие определяемых веществ заведомо известно. Например, установлено, чтоприсутствие спектральной линии с длиной волны 350,11 нм в эмиссионном спектре сплава, при возбуждении спектра электрической дугой, свидетельствует о наличии в сплаве бария; посинение водного раствора при добавлении к нему крахмала является АС на присутствие в нем I2 и наоборот.
Качественный химический анализ базируется на системе химических реакций, характерных для данного вещества - разделения, отделения иобнаружения.
К химическим реакциям в качественном анализе предъявляют следующие требования.
1. Реакция должна протекать практически мгновенно.
2. Реакция должна быть необратимой.
3. Реакция должна сопровождаться внешним эффектом (АС):
а) изменением окраски раствора;
б) образованием или растворением осадка;
в) выделением газообразных веществ;
г) окрашиванием пламени и.
Фотоэлектронная спектроскопия(ФЭС) – это наиболее широко используемый метод для изучения электронной структуры заполненных состояний на поверхности и в приповерхностной области (глубина 20-40 Ǻ). Физической основой метода служит фотоэлектрический эффект, в котором электрон, первоначально находящийся в состоянии с энергией связи Ев , поглощает фотон с энергией hν и покидает твердое тело с кинетической энергией:
Рис.1. Зонная диаграмма
Для того, чтобы зарегистрировать фотоэлектрон, должны быть выполнены следующие условия:
· Энергия фотона должна быть достаточна, чтобы электрон смог покинуть твердое тело, то есть hν ≥ Ев + Фо ()
· Скорость электрона должна быть направлена в сторону внешней поверхности.
· Электрон не должен потерять энергию в столкновениях с другими электронами на своем пути к поверхности.
Диапазон энергий фотонов, используемый в материаловедении ,простирается от ультрафиолета (УФ) до рентгеновского излучения. Практически энергетический диапазон простирается от 10 эВ, что близко к энергии связи электрона в атоме водорода (13,6 эВ), до энергий около 100 кэВ. При этих энергиях фотоны могут, проникая в твердое тело, взаимодействовать с электронами внутренних оболочек. Фотоны низких энергий используются для исследования спектров излучения в видимой области, связанного с далеко расположенными, более слабо связанными электронами. Эти внешние электроны участвуют в образовании химических связей, поэтому они не связаны с отдельными атомами и, следовательно, непригодны для элементного анализа.
В зависимости от энергии фотонов (длины волны), используемых для возбуждения электронов, фотоэлектронная спектроскопия обычно подразделяется на два типа:
· УФЭС (ультрафиолетовая фотоэлектронная спектроскопия), в которой используются фотоны ультрафиолетового спектрального диапазона 10-50 эВ (соответствующие длины волн от 1000 до 250А). В результате УФЭС используется для изучения валентной зоны и зоны проводимости.
· РФЭС (рентгеновская фотоэлектронная спектроскопия), в которой используется рентгеновское излучение с энергией квантов в диапазоне 100 эВ-10 кэВ (соответствующие длины волн от 100 до 1А). Как следствие РФЭС зондирует глубокие остовные уровни.
· Синхротронное излучение, в котором энегрии энергии фотонов от 40 до 1200 эВ. Позволяет исследовать остовные уровни и валентную зону.
Это разделение на два типа достаточно условно как с точки зрения объекта исследования (подразделение энергетических уровней на основные и валентные само по себе условно), так и с точки зрения используемых источников излучения (при использовании синхротронного излучения можно излучать фотоэмиссию от мягкого ультрафиолетового излучения до жесткого рентгеновского). Более того в обоих методах используются одни и те же физические процессы.
1 – источник рентгеновского (ультрафиолетового) излучения
4 – детектор электронов
Из уравнения h ν = Eсв + Eкин видно, что если известны величины h ν и Eкин , то можно определить энергию ионизации Eион или энергию связи Eсв соответствующего уровня. Для определения Eкин используются специально созданные электронные спектрометры. Электронный спектрометр (рис. 3) состоит из трех основных частей, в которых соответственно происходят генерация электронов, анализ их энергии и регистрация. В спектрометре поток ионизирующего излучения направляется на образец. Электроны могут быть выбиты из любой оболочки молекулы, ионизационный потенциал которой меньше энергии облучения. Выбитые электроны попадают в анализатор энергий электронов спектрометра. В анализаторе электроны описывают различные траектории в зависимости от своих энергий и напряжения, приложенного к электродам анализатора.
Наиболее распространен и хорошо известен сферический анализатор. В этом анализаторе к обкладкам сферического конденсатора прикладывается заданное напряжение. Разность потенциалов между двумя пластинами сферического конденсатора непосредственно связана с кинетической энергией электронов, прошедших через анализатор соотношением
где Eкин – кинетическая энергия электрона, V – разность потенциалов между двумя сферами радиусов R1 и R2 . Если менять напряжение на обкладках, то можно проанализировать спектр энергии Eкин , которой обладают электроны, падающие на входную щель анализатора.
Исследуемое вещество облучают монохроматическим рентгеновским излучением. В качестве источника рентгеновских монохроматических квантов может быть использована обычная рентгеновская трубка, анод которой сделан из материала, имеющего достаточно интенсивную и узкую линию рентгеновского излучения. В качестве таких линий выступают обычно – линия Mg (hν = 1253,6 эВ) или -линия Al (hν = 1486,6 эВ).
В качестве детектора электронов может быть использован обычный электрометр или пропорциональный счетчик. В серийных промышленных спектрометрах применяются электронные умножители. В этих приборах каждый попавший в них фотоэлектрон выбивает лавину вторичных электронов, которые регистрируются электронной схемой как отдельный импульс. Интенсивность рентгеноэлектронной линии определяется числом импульсов в единицу времени.
В фотоэлектронной спектроскопии твердых тел анализируется кинетическая энергия электронов, испущенных при облучении твердых тел моноэнергетическими фотонами с энергией hν
где I – энергия связи(ионизации) атомного или молекулярного уровня системы. Энергия фотонов известна, кинетическая энергия фотоэлектрона Еkin регистрируется с помощью спектрометра, а работа выхода спектрометра легко определяется с помощью калибровочных экспериментов.
Значит легко можно определить энергию связи (ионизации) соответствующего электронного уровня, которая зависит от характера распределения электронов в исследуемой системе.

Отсюда видно, что спектр фотоэмиссии I(E) это своего рода отпечаток плотности заполненных состояний исследуемого материала.
Фотоэлектронный спектр натрия
На рис. 5 проиллюстрирован процесс рентгеновской фотоэмиссии натрия. Пики соответствуют энергиям характеристических электронов, покидающих твердое тело без процессов, приводящих к потерям энергии.

Рис. 5. Энергетический спектр электронов образца Na, при синхротронном облучении фотонами с энергией 100 эВ. На энергетической шкале приведена энергия связи, т. е. hv — Етн.
Пики соответствуют энергиям характеристических электронов, покидающих твердое тело без процессов, приводящих к потерям энергии. Хвосты со стороны большей энергии связи соответствуют электронам, претерпевшим неупругое рассеяние и потерю энергии на пути из образца и выходящим поэтому с меньшей кинетической энергией, что приводит к кажущемуся возрастанию энергии связи.
Линии 2s и 2p отчетливо видны в виде острых пиков, это и есть остовные уровни, положение которых определяется энергией связи электронов, что является характеристикой данного элемента. Т.е. присутствие пиков при данной энергии связи является свидетельством присутствия на поверхности данного элемента (содержит информацию о химическом составе поверхности).
Сравнивая энергии пиков на экспериментальном спектре с известными энергиями связи в элементах можно выяснить какие элементы присутствуют в данном материале.
Из измеренных амплитуд пиков на спектре РФЕС, можно определить концентрацию элементов, из которых состоит поверхность.
Хочу отметить, что в общем случае вероятность фотоэмиссии максимальна при энергии фотонов близкой к порогу ионизации и она быстро уменьшается, если энергия фотонов значительно превосходит энергию связи электронов. Поэтому РФЭС – это метод для исследования в основном глубоких остовных уровней. Для исследования валентной зоны нужна меньшая энергия и использование УФ источника возбуждения.
Энергии 100эВ недостаточна для вырывания электронов из K-оболочек Na, но достаточна для создания вакансий в L-оболочках.
Химический сдвиг
Рис. 6. Химический сдвиг энергии связи линии Si2p в кремнии и SiO2

Рис. 7. Рентгеноэлектронный C1s-спектр этилового эфира трифторуксусной кислоты
Для иллюстрации зависимости энергии связи электрона в атоме от химического окружения атома часто используют рентгеноэлектронный 1s-спектр углерода этилового эфира трифторуксусной кислоты (рис. 3). Четыре максимума C1s почти равной интенсивности в весьма изящной форме представляют четыре окружения атомов углерода в этой молекуле.
В табл. 2 приведены значения энергий связи для 2p-уровня серы в газообразных соединениях. Из табл. 2 видно, что изменения энергии связи внутренних электронов могут достигать очень значительных величин в ряду соединений этого элемента.
Структура молекул
В настоящее время проведены многочисленные исследования, показывающие эффективность применения рентгеноэлектронной спектроскопии для решения различных вопросов структурной химии органических и неорганических соединений. Применение РФЭС в структурной химии можно показать на примере исследования 1s-спектров азота (N1s) в Na2 N2 O3 . До применения РФЭС предполагались три возможные структуры иона оксигипонитрата:
Рентгеноэлектронный спектр Na2 N2 O3 ясно указывает наличие структурно неэквивалентных атомов азота, и это исключает симметричную структуру (I). В то же время можно также ожидать, что структуры II и III будут давать две полосы в спектре N1s. Окончательный выбор между структурами (II) и (III) возможен только при анализе величины расщепления N1s полосы, связанной с различием величины электронной плотности на атомах азота.
Степень окисления
Рентгеноэлектронные спектры позволяют четко показать, что энергия связи внутреннего уровня атома в сильной степени зависит от степени окисления элемента, спектр которого изучается. Так, уже в первых работах было установлено, что при одинаковых ближайших соседях сдвиг внутренних уровней исследуемого атома в сторону увеличения Есв тем больше, чем больше степень окисления элемента в соединении.
При изучении поверхности металлов и сплавов часто возникают вопросы, является ли поверхность окисленной и какой именно компонент сплава окислен. Рентгеноэлектронные спектры в большинстве случаев помогают решить эту задачу, поскольку энергия связи электрона в металле обычно на несколько электронвольт меньше, чем в оксиде, причем с увеличением степени окисления также растет положительный химический сдвиг.

Интересный эксперимент описан в книге К. Зигбана с сотрудниками. Металлический бериллий испарялся при давлении 10 - 4 торр и осаждался на алюминиевую подложку. Образец облучали рентгеновскими квантами Kα -линии Al и изучали выбитые 1s-электроны с целью определения энергии связи 1s-электрона в бериллии. Спектр (рис. 4) состоит из двух линий одинаковой интенсивности, расстояние между которыми равно 2,9 ± 0,1 эВ. Появление двух линий вместо одной можно объяснить тем, что металл частично окислен и одна из линий электронного спектра соответствует металлическому, а другая – окисленному бериллию. Для проверки этого предположения образец нагревали на воздухе до полного окисления бериллия и снова снимался электронный спектр. На этот раз была получена только одна 1s-линия. Ее положение совпадает с положением линии меньшей кинетической энергии на рис. 4. Следовательно, это линия бериллия в окисле. Для дополнительного доказательства бериллиевый образец, полученный испарением в вакууме, частично восстанавливали цирконием, после чего был получен его электронный спектр. Хотя в спектре видны обе линии, линия, соответствующая окислу, гораздо менее интенсивна.
Рис. 8. Рентгеноэлектронные Be1s-спектр
Метод рентгеноэлектронной спектроскопии в настоящее время широко применяют для исследования поверхности твердых тел. В рентгеноэлектронной спектроскопии регистрируются электроны, вышедшие из слоя вещества, в котором они не успевают отдать часть своей кинетической энергии другим электронам и атомам в образце. Толщина этого слоя ~ 20 – 40 Ǻ, и, следовательно, рентгеноэлектронные спектры характеризуют только атомы поверхностного слоя. Вследствие этого рентгеноэлектронные спектры внутренних уровней атомов, входящих в соединение или материал, позволяют определять элементный состав поверхности, концентрацию элементов на поверхности, химическое состояние атомов на поверхности и приповерхностных слоях. Именно эти аналитические возможности метода позволяют изучать различные процессы, протекающие на поверхности.
Ультрафиолетовая фотоэлектронная спектроскопия.
Так как используются фотоны с низкой энергией происходит возбуждение только валентных уровней. Этот метод является инструментом изучения валентной полосы поверхности и её модификации в результате различных процессов на поверхности, таких как адсорбция, рост тонких пленок, химические реакции.
Для распределения плотности состояний в валентной зоне используется УФЭС с интегрированием по углам, которое в идеальном случае детектирует все фотоэлектроны, испускаемые над поверхностью образца.
Так же УФЭС с угловым разрешением позволяет определить закон дисперсии поверхностных состояний. Зависимость энергий связи фотоэлектронных спектров от угла выхода фотоэлектронов.
- Для учеников 1-11 классов и дошкольников
- Бесплатные сертификаты учителям и участникам
Реферат по теме
Определение понятия УФ спектроскопия …………………………………….3
Устройство спектрометров……………………….………………. …. ………10
Применение УФ спектроскопии….………. ………………………………..…14
Список использованных источников…….………………………………….16
Определение понятия УФ спектроскопия
Электронные спектры поглощения наблюдаются в результате поглощения ультрафиолетового и видимого излучения; при этом происходит переход (возбуждение) валентного электрона с занимаемого им уровня на уровень с более высокой энергией. По типу поглощаемого излучения электронную спектроскопию часто называют спектроскопией в ультрафиолетовой и видимой области, или УФ-спектроскопией.
При воздействии электромагнитного излучения на вещество происходит поглощение этого излучения. Поглощение в ультрафиолетовой и видимой областях спектра связано с возбуждением электронов. В этом случае говорят об электронных спектрах поглощения.
Спектр поглощения - это распределение по длинам волн (или частотам) интенсивности электромагнитного излучения при прохождении его через исследуемое вещество. На практике спектр поглощения получается следующим образом. Исследуемое вещество помещают между источником и приемником излучения. Источник с помощью специальных устройств посылает излучение с определенной или меняющейся длиной волны. Приемник измеряет интенсивность излучения, прошедшего через образец, и регистрирует его на ленте самописца. УФ-излучение является достаточно мощным (вспомните его воздействие на пигменты кожи при загаре или как выцветают на ярком солнечном свету многие красители). Под действием УФ света происходит возбуждение молекулы, то есть переход электронов на более высокий энергетический уровень и перераспределение электронной плотности в молекуле.
Необходимые для исследования количества вещества невелики — около 0,1 мг. В связи с этим УФ-спектроскопия является одним из наиболее распространенных физико-химических методов исследования органических и неорганических соединений.
Ультрафиолетовая (УФ-) спектроскопия позволяет исследовать взаимодействие ультрафиолетового излучения с электронным облаком молекул. Для аналитических целей используется диапазон ультрафиолетового излучения в пределах 10-8 – 4×10-7 м.
Так как энергия квантов ультрафиолетового излучения близка к энергии электронов, то при облучении молекул происходит возбуждение электронов и они переходят на разрыхляющие (нестабильные) молекулярные орбитали с более высокой энергией. На диаграмме (рис. 3.4) показаны переходы s-, n- и p-электронов на разрыхляющие орбитали (где n-переходы неподеленных пар гетероатомов).

Рис. 3.4. Переходы s-, n- и p-электронов на разрыхляющие орбитали
Через s®s* обозначается переход s-электронов со связующей на разрыхляющую s*-орбиталъ. s-Электроны – это гибридизованные электроны, участвующие в образовании простых С–С- и С–Н-связей. Они близко расположены к ядру, прочно с ним связаны, и для их перехода на разрыхляющую s-орбиталь необходима большая энергия. Такой энергией обладают ультрафиолетовые лучи с длиной волны менее 160 нм (труднодоступная область ультрафиолетового излучения – вакуумный ультрафиолет). Так как в образовании связей в молекулах алканов и нафтенов участвуют только s-электроны, то ультрафиолетовые спектры для этих углеводородов не характерны.
Через p®p* обозначается переход p-электронов двойных связей непредельных и ароматических углеводородов на разрыхляющие p*-орбитали; p-электроны слабее, чем s-электроны, связаны с ядром, и для их возбуждения и перехода на разрыхляющие орбитали требуется меньшая энергия. Так, олефины поглощают ультрафиолетовое излучение в области 170 – 180 нм, а диены и ароматические углеводороды - в области еще более длинных волн (> 200 нм). Таким образом, диеновые и ароматические углеводороды дают характерные ультрафиолетовые спектры в области 200 – 400 нм.
В случае гетероатомных соединений нефти (кислородные, сернистые, азотистые соединения) алифатического ряда УФ-спектры возникают в результате n®s* перехода неподеленных пар электронов гетероатомов. Полосы поглощения для гетероатомных соединений нефти находятся в коротковолновой области.
Если гетероатом находится рядом с двойной связью или ароматическим ядром, то поглощение происходит в области 200 – 300 нм (переход n®p*).
Энергия квантов ультрафиолетового излучения в 20 раз больше энергии квантов инфракрасного излучения и приближается к величине энергии, необходимой для разрыва связей в молекулах органических соединений. Поэтому ультрафиолетовое излучение сильно возбуждает молекулы органических соединений и ультрафиолетовые спектры представляют собой довольно грубую картину в отличие от тонкой структуры инфракрасных спектров.
В УФ-области поглощают все органические вещества. Длины волн менее 190 нм (дальняя или вакуумная область УФ-спектра) малопригодны для работы, так как в этой области поглощают компоненты воздуха — кислород и азот. Приборы для исследований в интервале длин волн 120-190 нм с вакуумными камерами существуют, однако они сложны и редко используются в обычной лабораторной практике. Для волн длиной более 200 нм воздух прозрачен, что делает ближнюю ультрафиолетовую и видимую области спектра (190-800 нм) удобными для измерений. В том же интервале прозрачен кварц, который в УФ-спектроскопии применяется как оптический материал для изготовления призм и кювет. Приборы для получения спектров поглощения в этой области просты и доступны. Необходимые для исследования массы вещества невелики — около 0,1 мг. В связи с этим УФ-спекгроскопия в настоящее время является одним из наиболее распространенных физико-химических методов исследования органических соединений.

Рис. 2.2. Энергетическая диаграмма молекулярных орбиталей, образующихся при сопряжении двух двойных связей.
Группировки, вызывающие избирательное поглощение электромагнитного колебания в УФ-области, называются хромофорами. Основными хромофорами, дающими максимум поглощения в области 200-800 нм, являются системы сопряженных двойных связей. Орбитали, образуемые двумя сопряженными двойными связями, представлены на рис. 2.2. Видно, что при взаимодействии двух л-орбиталей, соответствующих изолированным двойным связям, образуются две новые орбитали: связывающая (я+л) и разрыхляющая (л-л). Возбужденному состоянию соответствуют две аналогичные орбитали. Следовательно, для возбуждения электронов сопряженной системы, т.е. для осуществления перехода с высшей занятой (л-л) на низшую свободную (я*+ я*) орбиталь, требуется меньше энергии, чем для возбуждения электронов изолированных двойных связей (п-*п*). Ясно, что сопряженные двойные связи будут поглощать кванты света с большей длиной волны, чем изолированные двойные связи.
С ростом числа таких сопряженных связей энергия, необходимая для возбуждения электронов, уменьшается, и поглощение света будет наблюдаться при больших длинах волн. В ароматических системах переход электрона в возбужденное состояние осуществляется также при меньшей затрате энергии, чем в случае наличия изолированной двойной связи. Таким образом, основными хромофорами в УФ-спектроскопии являются сопряженные С=С-связи, карбонильная группа С=0, системы С=С-С=0 и ароматическое ядро.
УФ-спектр органического вещества характеристичен, так как поглощение определяется только собственно хромофором и его ближайшим окружением, т.е. один и тот же хромофор проявляется практически одинаково как в простых, так и в самых сложных молекулах. В зависимости от непосредственного окружения одной и той же хромофорной группировки положение максимума поглощения в УФ-спектрах различных соединений может несколько изменяться. Сдвиг максимума в сторону более длинных волн принято называть батохромным, а сдвиг в сторону более коротких волн — гипсохромным. Интенсивность поглощения в спектре связана с вероятностью данного типа электронного перехода. Однако далеко не все переходы, формально кажущиеся возможными, осуществляются в действительности. Есть правила отбора, определяющие разрешенные и запрещенные переходы. Эти правила учитывают в основном симметрию молекулы, а также электронную симметрию основного и возбужденного состояний. Запрещены переходы, при которых происходит изменение спина электрона. Интенсивность поглощения, соответствующего разрешенным переходам, обычно высока, молярный коэффициент поглощения достигает тысяч, а иногда и сотен тысяч единиц, тогда как для запрещенных переходов значение е составляет десятки, реже — сотни единиц.
Устройство спектрометров

Спектрометры для получения УФ-спектров имеют следующее устройство. В качестве источника УФ-излучения обычно применяется водородная лампа (электрическая дуга в атмосфере водорода при низком давлении), которая дает практически непрерывный спектр излучения в области 190-360 нм. Для работы в видимой области служит лампа накаливания с вольфрамовой спиралью. Излучение от источника попадает в монохроматор, состоящий из зеркала, кварцевой призмы и щели. Отражаясь от зеркала, свет разлагается призмой и затем с помощью щели из спектра выделяется узкая область. При вращении призмы спектр перемещается по отношению к щели, что позволяет получать лучи света со строго определенной длиной волны, обычно с точностью ±0,5 нм. Монохроматическое излучение пропускается через кварцевую кювету, содержащую раствор исследуемого вещества в прозрачном для УФ-области растворителе. Толщина кювет 1-10 см, наиболее распространенные кюветы имеют сечение 1x1 см, и для их заполнения требуется около 3 мл раствора. Интенсивность прошедшего через кювету света измеряется с помощью фотоэлемента, величина тока которого пропорциональна интенсивности падающего света. Ток усиливается и регистрируется потенциометром. Сравнивается интенсивность светового луча, прошедшего через исследуемый раствор, и луча, пропущенного через аналогичную кювету с чистым растворителем. Получаемая разность соответствует поглощению растворенного исследуемого вещества. В современных регистрирующих приборах световой поток делится на два одинаковых пучка, один из которых проходит через исследуемый раствор, а другой — через растворитель, причем как сравнение интенсивностей прошедших через кюветы световых потоков, так и непрерывное изменение длин волн производится автоматически. В том и другом случае получают УФ-спектр вещества, представляющий собой зависимость оптической плотности раствора D (или е) от длины волны поглощаемого света. В точках максимума мольный коэффициент поглощения вычисляют по уравнению:

Рис. 2.3. УФ-спектр никлопентадиена
Обычно УФ-спектр характеризуют длиной волны, при которой наблюдается максимум поглощения, и мольным коэффициентом поглощения в этом максимуме. Например, спектр циклопентадиена (см. рис. 2.3) достаточно точно может быть передан записью: ХмакС(в гексане) = 240 нм (е = 3400 л/моль-см). УФ-спектр вещества может иметь несколько максимумов поглощения, каждый из которых соответствует различным типам электронных переходов. В этом случае при цифровой записи спектра перечисляются длины волн максимумов поглощения и в скобках приводятся значения е, соответствующие данному максимуму. Если спектр имеет относительно сложный контур (например, тонкая структура какого-либо из максимумов), целесообразно приводить непосредственно рисунок спектра, так как часто его специфические особенности характеристичны и позволяют делать определенные выводы.
Мольный коэффициент экстинкции для каждого поглощающего в УФ-области вещества при данной длине волны в одном растворителе имеет строго постоянное значение, определяющее оптическую плотность раствора при заданной концентрации. Точность измерения оптической плотности весьма высока — погрешность измерений составляет, в зависимости от конструкции прибора, ±0,2* 1% от определяемой величины. Следовательно, измеряя оптическую плотность раствора, при известной ? можно с высокой точностью определять концентрацию вещества. Измерения не обязательно проводить при длине волны, соответствующей максимуму поглощения. Таким образом, УФ-спектроскопия позволяет с достаточно высокой степенью точности осуществлять количественный анализ растворов и в том числе наблюдать изменение концентрации вещества во времени. Последнее находит широкое применение при исследовании кинетики химических реакций.
В качестве растворителей в УФ-спектроскопии используются алканы (гексан, гептан), этанол, вода, реже — диоксан. Концентрации исследуемых растворов (обычно 10 -10"6 моль/л) подбираются так, чтобы их оптическая плотность находилась в пределах 0,3-0,7. Это обеспечивает максимальную точность измерений. Для получения точных значений столь малых концентраций используется метод последовательных разведений, при использовании которого взвешивают раствор и растворитель. В данном случае предполагается совершение минимального количества ошибок, чем при разведении по объему.
Необходима особо тщательная очистка применяемых растворителей, так как примесь, например, ароматических соединений в количестве КГ5 моль/л делает алканы непригодными для работы. Методы очистки растворителей хорошо отработаны. Наиболее просто (перегонкой) очищается вода — самый доступный растворитель. Замена растворителя в отдельных случаях может вызвать некоторые изменения как в положении полос (на 2-10 нм), так и в их интенсивности (на 10-20%). Как правило, такая замена влияет на спектры полярных веществ и практически не сказывается на УФ-спектрах неполярных соединений. Наиболее сильные изменения в спектрах обусловлены химическим взаимодействием вещества с растворителем (в частности, образованием водородной связи), а также изменением степени диссоциации или соотношения таутомерных форм вещества. Во всех таких случаях следует проверить, выполняется ли для данного раствора закон Бугера- Ламберта-Бера.
Таким образом, УФ-спектроскопия позволяет определить в исследуемых соединениях группировки хромофоры и дает возможность проведения количественного анализа веществ, содержащих такие группировки. Этот метод находит широкое применение не только в лабораторной практике, но и в химической и пищевой промышленности, например для определения стирола в его смесях с ди- винилбензолом, определения каротиноидов, бензопирена и т.д.
Применение УФ спектроскопии
В настоящее время для структурного анализа органических соединений электронная спектроскопия имеет ограниченное применение. Гораздо более важная область использование УФ-спектроскопии – это количественный анализ. Использование данного метода эффективно как в случае изучения кинетики реакции, так и при определении примесей в образце органического вещества. Соединения, поглощающие в УФ-области с большой интенсивностью, часто могут быть определены даже при низкой концентрации, если они присутствуют в качестве примесей в образцах веществ, имеющих слабое поглощение в области max примеси. Классическим примером является определение бензола, присутствующего в низкой концентрации в качестве примеси в этиловом спирте.
Чтобы провести количественное определение вещества спектрофотометрическим методом, необходимо на основании снятого спектра измерить интенсивность поглощения света этим веществом при выбранной длине волны. Однако это возможно лишь в тех случаях, когда установлено, что в интервале возможных концентраций поглощение подчиняется основному закону светопоглощения. Теоретически концентрацию можно определить при любой длине волны. В то же время следует подчеркнуть, что минимальная ошибка определения получается при тех длинах волн, которые отвечают следующим требованиям:
1) выбранная полоса должна быть по возможности свободна от наложения полос поглощения других компонентов анализируемой системы;
2) выбранная полоса должна обладать достаточно высоким коэффициентом поглощения для индивидуального соединения.
Такие полосы называются аналитическими. При анализе используют максимум или минимум полосы поглощения и не следует производить измерения на участках крутого спада или подъема кривой.
Как структурно-аналитический метод УФ-спектроскопия значительно менее информативна по сравнению с другими и носит в основном эмпирический характер, поскольку зависимость между характером поглощения и структурой молекулы не имеет строгого физико-математического обоснования, что, однако, не мешает широкому его использованию.
РЕФЕРАТ ПО ХИМИИ ФОТОЭЛЕКТРОННАЯ СПЕКТРОСКОПИЯ.docx
Физические основы метода ………………………………………….…………6
Общая характеристика фотоэлектронных спектрометров …………………..13
В настоящее время нет общепринятой классификации многочисленных физических методов изучения состава, строения и свойств молекул, твердых тел и поверхности. Все методы можно классифицировать, во-первых, по характеру взаимодействия излучения или потока частиц с веществом, во-вторых, по изучаемым параметрам вещества, как принято в учебнике Л.В. Вилкова и Ю.А. Пентина [1, 2]. Если рассматривать только методы, ориентированные на изучение строения вещества, можно говорить о методах исследования геометрического строения и методах исследования электронной структуры. К первой группе кроме рентгеновских дифракционных методов и газовой электронографии можно отнести методы колебательной спектроскопии и ядерного магнитного резонанса, а во второй группе наиболее информативны методы фотоэлектронной спектроскопии и рентгеновской флуоресцентной спектроскопии 5. Безусловно, такое деление методов на две группы не является строгим, так как структура электронных оболочек зависит от геометрии ядерного остова, а межатомные расстояния и углы между связями определяются электронами атомов, образующих химическое соединение. Поэтому при таком делении методов на две группы учитывается преимущественная информация, которую можно получить данным методом.
До появления метода фотоэлектронной спектроскопии, основанного на явлении внешнего фотоэффекта, экспериментальную информацию об электронном строении атомов и молекул в основном получали из оптических спектров и спектров неупругого рассеяния электронов. Действительно, трудно переоценить роль обнаруженных серий в спектрах атома водорода, щелочных металлов и результатов первых опытов Дж. Франка и Г. Герца по рассеянию электронов на атомах в развитии современных квантово- механических представлений о строении вещества. Но энергии возбуждения атомов и молекул, полученные методом оптической спектроскопии зависят как от заполненных, так и от вакантных орбиталей, что затрудняет получение информации о заполненных орбиталях. К началу 60-х годов экспериментальные данные об энергиях связи валентных электронов многоатомных молекул, как правило, исчерпывались двумя или тремя уровнями p-типа. Отсутствие экспериментальных данных о многоцентровых молекулярных орбиталях (МО) s-типа вместе с простотой и наглядностью метода локализованных гибридных орбиталей обусловили преимущественное использование последнего метода в описании электронной структуры и химических связей молекул. Более того, в изданных в 1997 году учебниках химии для средней школы делокализация МО допускается только для p-электронов, тогда как остальные электронные пары принимаются локализованными на атомах или связях. Такое описание электронной структуры молекул без упоминания о существовании альтернативного метода многоцентровых МО, адекватно описывающего свойства молекул, нельзя признать целесообразным.
ФИЗИЧЕСКИЕ ОСНОВЫ МЕТОДА
Уравнение Эйнштейна для ионизации свободных атомов имеет вид
где I1 - первый потенциал ионизации, Ti - энергия электронного возбуждения иона, Екин и Еи - кинетическая энергия фотоэлектрона и энергия отдачи иона. Большое различие в массах электрона и иона позволяет последним слагаемым в (1) пренебречь и записать уравнение в привычной для фотоэффекта форме
где Ii = I1 + Ti , то есть потенциал ионизации, отвечающий переходу атома в i-е электронное состояние иона. Здесь необходимо договориться об использовании терминов "энергия ионизации" и "потенциал ионизации". Из методики измерения энергии, необходимой для удаления наименее связанного электрона, до наших дней дошел второй термин, тогда как для энергии связи внутренних электронов в методе РФЭС используется первый термин. Ниже оба термина будем считать равнозначными для всех электронов.
Для молекул необходимо учитывать изменение в процессе ионизации колебательной (Gv) и вращательной (Ft) энергии:
В акте поглощения фотона и эмиссии электрона вращательное состояние ядерного остова изменяется незначительно (Ft ≤ 0,02 эВ), что позволяет упростить выражение (3):
где Iai - энергия адиабатического перехода в i-е электронное состояние, то есть переход без изменения колебательной энергии; Ivi - энергия перехода молекулы в произвольное электронно-колебательное состояние. Энергию перехода без изменения межъядерных расстояний, фактически соответствующего переходу в максимально заселенное колебательное состояние иона, принято называть вертикальным потенциалом ионизации. Поскольку в каждом эксперименте энергия фотона монохроматического излучения известна, для определения энергии ионизации в i-е электронное состояние достаточно измерить кинетическую энергию фотоэлектронов.
Какова же связь наблюдаемых энергий ионизации с электронной структурой? Значения Iai , по определению, для молекул равны разностям энергий электронных состояний иона Е и i и молекулы Е м 0 :
а значения вертикальных энергий ионизации - разностям полных энергий при совпадающей геометрии иона и молекулы:
Здесь Еi ( max ) - энергия иона на максимально заселенном колебательном уровне. В методе молекулярных орбиталей показано, что, если для иона использовать МО, рассчитанные для молекулы (приближение "замороженных" орбиталей), разность энергий иона и молекулы совпадает с энергией ионизованного уровня (теорема Купманса) :
Следовательно, можно говорить о расчетных и экспериментальных орбитальных энергиях ионизации. Первая полоса в ФЭ-спектре соответствует состоянию иона с вакансией на верхней МО, вторая - состоянию с вакансией на следующей орбитали и т.д. (рис. 1). Пусть энергия фотона достаточна для выбивания электрона с одной из трех верхних орбиталей. Тогда в спектре наблюдаются три полосы, значения Iвi здесь совпадают с энергиями МО ξi :
В приближении "замороженных" орбиталей существует взаимно однозначное соответствие между заполненными МО и полосами в ФЭ-спектрах.
Интерпретацию ФЭ-спектров валентных и остовных уровней на основе идеи о "замороженных" орбиталях следует рассматривать как приближенную по двум причинам. Во-первых, равенство Iвi = - ξi нарушается перестройкой электронной оболочки, вызванной появлением электронной вакансии. В результате релаксации электронов орбитальные энергии в ионе изменяются, а энергия ионизации снижается (Iвi моментов неспаренных электронов (спин-орбитальное расщепление);
2) межэлектронное взаимодействие для атомов и молекул, имеющих в исходном состоянии частично заполненные орбитали (например, О2 , NО);
3) электронно-колебательные взаимодействия в вырожденных электронных состояниях.
Колебательная структура в ФЭ-спектрах наблюдается для двухатомных и простых многоатомных молекул. Колебательные контуры полос и частоты нормальных колебаний в сравнении с частотами невозбужденных молекул позволяют установить связывающий характер той орбитали, с которой удаляется электрон. Рассмотрим на примере двухатомной молекулы наиболее характерные типы колебательной структуры полос.
В нижнем состоянии иона (рис. 2) равновесное межатомное расстояние r' е совпадает с исходным равновесным расстоянием основного состояния молекулы r''е , поэтому интеграл перекрывания колебательных функций нижнего и верхнего состояний, определяющий вероятность перехода, Rυ’υ’’ = [ ψ''0 (Q) ׀ ψ' (Q)] , близок к единице для υ' = 0. Такое распределение интенсивности характерно для ионизации при удалении электрона с несвязывающей, слабосвязывающей или слабоантисвязывающей орбиталей. При удалении связывающего электрона равновесное межатомное расстояние, как правило, возрастает ( r'e > r''e), поэтому вертикальный, наиболее вероятный переход уже не совпадает с адиабатическим (состояния ˜A и ˜С на рис. 2). Частота колебаний иона при этом может снизиться по сравнению с частотой колебаний молекулы в полтора-два раза. Удаление электрона с антисвязывающей МО (состояние ˜В) ведет к возрастанию частоты колебания. Отсутствие колебательной структуры в полосе при разрешении спектрометра ΔE ионизации всех или значительной части электронных уровней;
2) разрешение в спектрах должно обеспечивать анализ колебательной структуры полос (ΔE ≤ 0,1 эВ);
3) вакуум в энергоанализаторе электронов не должен превышать 10 - 6 мм рт.ст.
Схема одной из модификаций фотоэлектронного спектрометра, предназначенного для изучения газов и паров веществ, приведена на рис. 5. К основным узлам и системам спектрометра можно отнести источник фотонов, ионизационную кювету, энергоанализатор электронов, детектор электронов, вакуумную систему и систему управления спектрометром и обработки информации. Отличительной особенностью спектрометров с источниками излучения в области вакуумного ультрафиолета является отсутствие каких-либо окон между источником излучения (разряд в потоке рабочего газа), ионизационной кюветой, анализатором и детектором электронов.
Ультрафиолетовая (УФ-) спектроскопия позволяет исследовать взаимодействие ультрафиолетового излучения с электронным облаком молекул. Для аналитических целей используется диапазон ультрафиолетового излучения
в пределах 10 -8 – 4×10 -7 м.
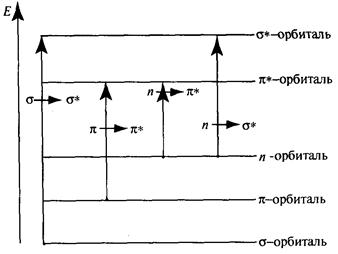
Так как энергия квантов ультрафиолетового излучения близка к энергии электронов, то при облучении молекул происходит возбуждение электронов и они переходят на разрыхляющие (нестабильные) молекулярные орбитали
с более высокой энергией. На диаграмме (рис. 3.4) показаны переходы s-, n- и p-электронов на разрыхляющие орбитали (где n-переходы неподеленных пар гетероатомов).
-
Рис. 3.4. Переходы s-, n- и p-электронов на разрыхляющие орбитали
Через s®s* обозначается переход s-электронов со связующей на разрыхляющую s*-орбиталъ. s-Электроны – это гибридизованные электроны, участвующие в образовании простых С–С- и С–Н-связей. Они близко расположены к ядру, прочно с ним связаны, и для их перехода на разрыхляющую s-орбиталь необходима большая энергия. Такой энергией обладают ультрафиолетовые лучи с длиной волны менее 160 нм (труднодоступная область ультрафиолетового излучения – вакуумный ультрафиолет). Так как в образовании связей в молекулах алканов и нафтенов участвуют только
s-электроны, то ультрафиолетовые спектры для этих углеводородов не характерны.
Через p®p* обозначается переход p-электронов двойных связей непредельных и ароматических углеводородов на разрыхляющие p*-орбитали;
p-электроны слабее, чем s-электроны, связаны с ядром, и для их возбуждения и перехода на разрыхляющие орбитали требуется меньшая энергия. Так, олефины поглощают ультрафиолетовое излучение в области 170 – 180 нм, а диены и ароматические углеводороды - в области еще более длинных волн (> 200 нм). Таким образом, диеновые и ароматические углеводороды дают характерные ультрафиолетовые спектры в области 200 – 400 нм.
В случае гетероатомных соединений нефти (кислородные, сернистые, азотистые соединения) алифатического ряда УФ-спектры возникают в результате n®s* перехода неподеленных пар электронов гетероатомов. Полосы поглощения для гетероатомных соединений нефти находятся в коротковолновой области.
Если гетероатом находится рядом с двойной связью или ароматическим ядром, то поглощение происходит в области 200 – 300 нм (переход n®p*).
Энергия квантов ультрафиолетового излучения в 20 раз больше энергии квантов инфракрасного излучения и приближается к величине энергии, необходимой для разрыва связей в молекулах органических соединений. Поэтому ультрафиолетовое излучение сильно возбуждает молекулы органических соединений и ультрафиолетовые спектры представляют собой довольно грубую картину в отличие от тонкой структуры инфракрасных спектров.
Ультрафиолетовые спектры записываются в форме зависимости логарифма молярного коэффициента поглощения lg e от длины волны l.Молярный коэффициент поглощения e является коэффициентом в уравнении Бугера – Ламберта – Бера. Кривая УФ-спектра имеет один или несколько максимумов поглощения, которые характеризуются соответствующими длинами волн lmах. Из рис. 3.5, на котором приведен УФ-спектр нафталина (1) и антрацена (2), видно, что lmах, соответствующие максимумам поглощения антрацена, смещены в длинноволновую часть спектра по сравнению с нафталином. Это объясняется тем, что в молекуле антрацена число сопряженных двойных связей больше, чем в молекуле нафталина. Известно, что с увеличением длины цепи сопряжения в молекуле облегчается переход p®p*. Так как для такого перехода требуется меньшая энергия, поглощается волна большей длины.
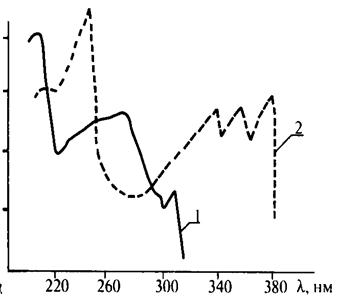
Рис. 3.5. УФ-спектр нафталина (1) и антрацена (2)
Введение алкильных групп в бензольное ядро вызывает сдвиг обеих полос поглощения для бензола в длинноволновую область спектра. Для смесей алкилбензолов максимум поглощения лежит в области 255 – 275 нм. Для алкилнафталинов максимум поглощения находится в области 275 – 290 нм; в области 310 – 330 нм имеются также два характерных максимума. Для алкилпроизводных антрацена и фенантрена характерны широкие полосы поглощения с несколькими максимумами в области 310 – 380 нм и 280 – 310 нм.
3.5.2. Масс-спектроскопия
В последние годы для структурных исследований молекул органических соединений используют масс-спектроскопию, основанную на ионизации молекул электронным ударом.
Масс-спектрометр – это прибор, который позволяет разделять ионы, полученные бомбардировкой молекул электронами: разделение ионов происходит по их массам. Схема прибора приведена на рис. 3.6.
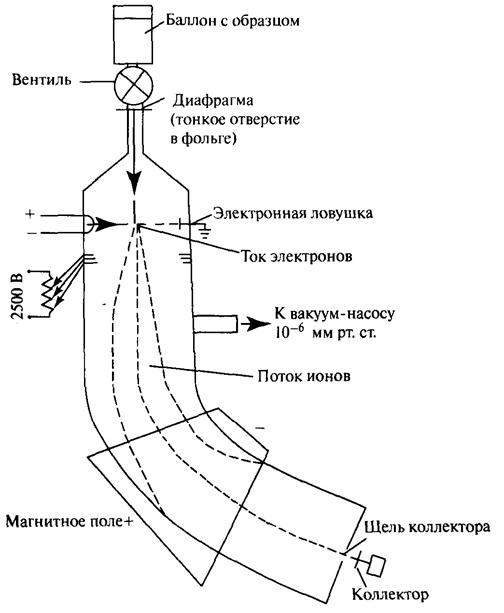
Рис. 3.6. Схема масс-спектрометра
Пары образца из баллона поступают через диафрагму в вакуумированную камеру (остаточное давление 10 -6 мм рт. ст.), где сталкиваются с потоком электронов, движущихся от катода (накаленная нить) к электронной ловушке (земля). Электроны выбивают из молекул орбитальные электроны
и превращают молекулы в ионы.
Кроме электроноударного метода ионизации молекул в химии нефти для высокомолекулярных нефтяных компонентов, обладающих низкой химической стабильностью, применяется метод полевой ионизации (ионизации под действием сильного неоднородного электрического поля). Развитием полевой ионизации является метод полевой десорбции: образец наносится на острие вольфрамового эмиттера, помещенного в сильное неоднородное электрическое поле, и эмиттер медленно нагревается электрическим током. Вследствие туннелирования наиболее подвижных электронов в эмиттер происходит ионизация молекул, и образующиеся молекулярные ионы десорбируются в газовую фазу. Ионы под действием возрастающего электрического поля, приложенного к сеткам ионной пушки, втягиваются в ионную пушку и ускоряются. Диаметр диафрагм сеток увеличивается по ходу движения ионов, поэтому ионы образуют расходящийся пучок, который попадает в магнитное поле. Нейтральные молекулы выводятся из камеры с помощью вакуумного насоса.
Изменяя напряженность магнитного поляили скорость движения ионов, можно поочередно фокусировать ионы различной массы на коллектор (с помощью щели ~0,1 мм). Образующийся при этом ионный ток усиливается и подается на записывающее устройство.
На рис. 3.7 приведен общий вид масс-спектрограммы. На оси абсцисс откладывается отношение массы иона m к его заряду z. Последний пик соответствует самому тяжелому молекулярному иону, который образуется при отщеплении от молекулы одного электрона. Остальные пики соответствуют осколочным ионам, образующимся при распаде молекулярного иона.
Рис. 3.7. Общий вид масс-спектрограммы
Рассмотрим упрощенную схему масс-спектрального распада изопропилбензола:
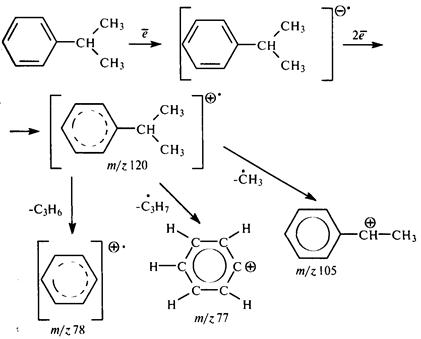
Молекула изопропилбензола под действием электронного удара превращается в анион-радикал, затем – в катион-радикал, распад которого приводит к положительным осколочным ионам различной массы.
С помощью масс-спектроскопии в химии нефти можно определить:
– молекулярную массу углеводорода или гетероатомного соединения;
– элементный состав вещества. Для этого с помощью приборов с высокой разрешающей способностью необходимо вычислить молекулярную массу вещества с точностью до 10 -3 – 10 -4 . По масс-спектру вещества определяют его структурную формулу, для чего следует идентифицировать осколочные ионы по их массам и по фрагментам молекулы воссоздать ее структуру;
– качественный и количественный состав углеводородных смесей.
Количественный состав простых смесей нескольких веществ можно установить на основании интенсивности пиков молекулярных ионов. На основании данных калибровочных графиков в координатах интенсивность пика – процентное содержание компонента по масс-спектрограмме смеси неизвестного состава определяют процентное содержание каждого компонента в смеси.
В последние годы при исследовании нефтей применяется метод хромато-масс-спектрометрии. Этот метод предполагает использование газового
хроматографа в блоке с масс-спектрометром и ЭВМ. В хроматографе происходит разделение углеводородов, которые затем поступают в масс-спектрометр; данные масс-спектра поступают в ЭВМ, которая расшифровывает спектр и идентифицирует углеводород.
Анион-радикал, образовавшийся в результате захвата электрона молекулой вещества (молекулярный ион) распадается с образованием радикалов, анионов и анион-радикалов.
В качестве примера рассмотрим схему масс-спектрального распада нефтяного сульфида (этилпропилсульфида):
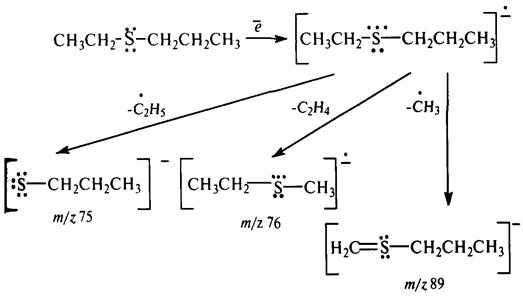
Углеводороды не поглощают электроны низкой энергии, поэтому масс-спектроскопическое исследование гетероатомных соединений можно проводить, не выделяя их из нефтяной фракции в чистом виде.
3.5.3. Спектроскопия ядерного магнитного резонанса
Кроме массы и заряда, ядро обладает третьей характеристикой – моментом количества движения, который обусловлен его вращением вокруг оси – спином. Поскольку ядро заряжено, его вращение вокруг собственной оси приводит к круговому движению заряда ядра, что формально аналогично круговому электрическому току (рис. 3.8).
Круговой ток создает магнитное поле, так что вращающееся ядро подобно крошечному магниту, ось которого совпадает с осью спина, поэтому ядро может характеризоваться магнитным дипольным моментом. Ядра, имеющие четный заряд (порядковый номер) и четное массовое число, не обладают магнитным моментом. У ядер, имеющих нечетный заряд, но четное массовое число, магнитный момент I = 1 (например, ядра азота). У ядер, имеющих нечетный заряд и нечетное массовое число, магнитный момент
I = 1/2 (ядра водорода, фтора, фосфора). Ядра изотопа углерода 13 С6 и изотопа кремния 29 Si14 также имеют нечетное массовое число и четный заряд.

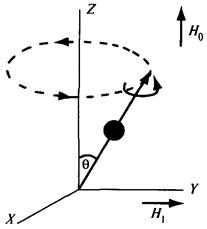
Рис. 3.8. Круговое движение Рис. 3.9. Движение ядра
заряда ядра в магнитном поле
Рассмотрим ядро, магнитный диполь которого ориентирован под некоторым углом Q к направлению силовых линий постоянного магнитного поля Н0 (рис. 3.9). Это поле обусловливает появление силы, стремящейся расположить ядро-магнит вдоль поля. Но, поскольку ядро вращается и обладает моментом количества движения, оно сопротивляется этому воздействию. В результате этого наблюдается прецессия магнита-ядра: кроме вращения вокруг своей оси, ядро вращается вокруг направления постоянного магнитного поля, подобно тому, как прецессирует волчок, если он наклонен по отношению к силовым линиям гравитационного поля Земли. Угловая скорость этой прецессии w (в рад/с) не зависит от угла Q, но зависит от напряженности постоянного магнитного поля H0:
где g – гиромагнитное отношение, в которое входит ядерный магнитный момент.
Рассмотрим влияние небольшого магнитного поля напряженностью Н1, перпендикулярного к постоянному магнитному полю Н0,на движение ядра в магнитном поле.Последнее стремится отклонить диполь (ось спина) в плоскость XY,однако действие поля незначительно. Если же поле Н1 начнет вращаться вокруг направления линий магнитного поля Н0 (т. е. если оно будет переменным), то, когда частота этого поля достигнет частоты прецессии ядра, произойдет поглощение ядром кванта энергии. Это поглощение называется резонансным.
Тот же самый результат может быть получен, если частота переменного поля остается постоянной, а изменяется напряженность постоянного магнитного поля. При изменении напряженности постоянного магнитного поля изменяется частота прецессии ядра, и, когда она достигает частоты переменного магнитного поля, происходит резонанс. Задача спектроскопии ядерного магнитного резонанса состоит в том, чтобы определить напряженность постоянного магнитного поля, при которой наступает резонанс ядер исследуемого образца в переменном поле определенной частоты n. В этом случае частота n равна частоте ядерного магнитного резонанса (ЯМР). В табл. 3.2 приведены значения ЯМР для ядер различных атомов. Из таблицы следует, что ядра кислорода и углерода, спин которых равен нулю, не являются магнитными, поэтому они не способны к ядерному магнитному резонансу.
Читайте также: