
Синдром луи бар реферат
Обновлено: 05.07.2024
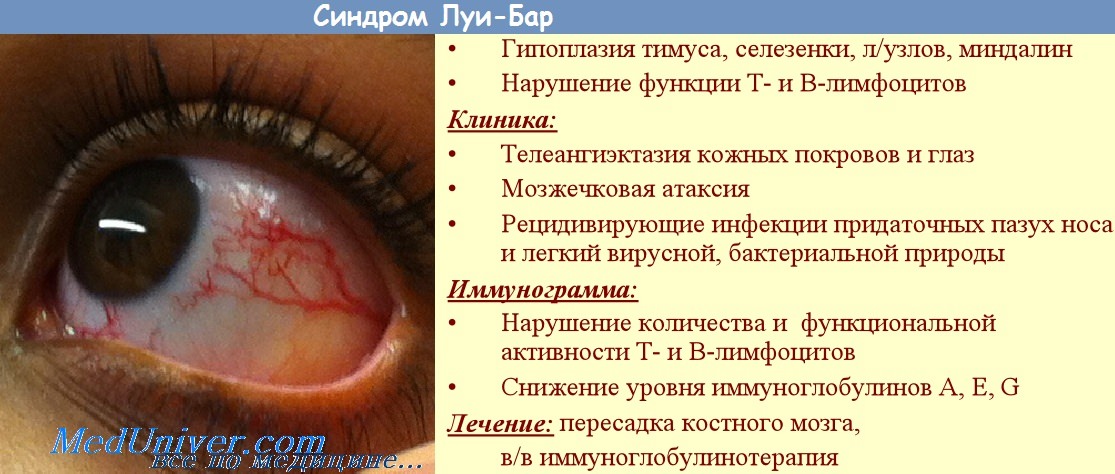
Наследственное заболевание, проявляющееся мозжечковой атаксией, телеангиэктазиями кожи и конъюнктивы глаз, недостаточностью Т-клеточного звена иммунитета. Последнее приводит к тому, что синдром Луи-Бар сопровождается частыми респираторными инфекциями и склонностью к возникновению злокачественных опухолей. Диагностируется синдром Луи-Бар на основании анамнеза и клинической картины заболевания, данных иммунограммы, результатов офтальмологического и отоларингологического обследования, МРТ головного мозга и рентгенографии легких. В настоящее время синдром Луи-Бар не имеет специфического и эффективного лечения.
Причины и патогенез
В основе патологических изменений, сопровождающих синдром Луи-Бар, лежат генетические нарушения, приводящие к развитию врожденной нейроэктодермальной дисплазии. Синдром Луи-Бар является аутосомно-рецессивным заболеванием, т. е. проявляется клинически только при получении рецессивного гена сразу от обоих родителей.
Морфологически атаксия-телеангиэктазия характеризуется дегенеративными изменениями тканей мозжечка, в частности потерей зернистых клеток и клеток Пуркинье. Дегенеративные изменения могут затрагивать зубчатое ядро мозжечка (nucleus dentatus), черную субстанцию (substantia nigra) и некоторые отделы коры головного мозга, иногда поражаются спиномозжечковые пути и задние столбы спинного мозга.
Синдром Луи-Бар сочетается с гипоплазией или аплазией тимуса, а также с врожденным дефицитом IgA и IgE. Эти нарушения в системе иммунитета приводят к появлению у пациентов частых инфекционных заболеваний, склонных к длительному и осложненному течению. Кроме того, иммунные нарушения могут потенцировать развитие злокачественных новообразований, зачастую берущих свое начало в структурах лимфоретикулярнойсистемы.
Клиника
Атаксия. Наиболее часто синдром Луи-Бар начинает проявляться клинически в возрасте от 5 месяцев до 3 лет. Во всех случаях заболевания синдром Луи-Бар манифестирует с появления мозжечковой атаксии, признаки которой становятся очевидными, когда ребенок начинает ходить. Наблюдаются нарушения равновесия и походки, дрожание во время двигательного акта (интенционный тремор), качание туловища и головы. Зачастую атаксия настолько выражена, что имеющий синдром Луи-Бар больной не может ходить. Мозжечковая атаксия сочетается с мозжечковой дизартрией, характеризующейся невнятной скандированной речью. Отмечается мышечная гипотония, снижение или полное исчезновение сухожильных рефлексов, нистагм, глазодвигательные нарушения и косоглазие.
Телеангиэктазии. В большинстве случаев появление сопровождающих синдром Луи-Бар телеангиэктазий происходит в возрасте от 3 до 6 лет. В некоторых случаях их возникновение отмечается в более поздний период и очень редко в течение первого месяца жизни. Телеангиэктазии (сосудистые звездочки) представляют собой имеющие различную форму красноватые или розовые пятнышки или разветвления. Они обусловлены расширением мелких сосудов кожи. Следует отметить, что телеангиэктазии могут быть проявлением многих других заболеваний (например, розацеа, СКВ, дерматомиозита, пигментной ксеродермы, хронического лучевого дерматита, мастоцитоза и пр.). Однако в сочетании с атаксией они дают специфическую для синдрома Луи-Бар клиническую картину.
Кожные проявления атаксии-телеангиэктазии могут включать появление веснушек и пятен цвета кофе с молоком, участков обесцвеченной кожи. Наличие гипо- и гиперпигментаций делает кожные симптомы синдрома Луи-Бар схожими с клиникой пойкилодермии. У многих больных отмечается сухость кожи и участки гиперкератоза. Может наблюдаться гипертрихоз, ранняя седина волос, кожные элементы, напоминающие акнеили проявления псориаза.
Инфекции дыхательных путей. Характеризующее синдром Луи-Бар поражение иммунной системы приводит к возникновению частых рецидивирующих инфекций дыхательных путей и уха: хронических ринитов, фарингитов, бронхитов, пневмоний, отитов, синуситов. Их особенностями являются: стертость границ между периодом обострения и ремиссии, скудность физикальных данных, плохая чувствительность к антибактериальной терапии и длительное течение. Каждая подобная инфекция может стать для больного атаксией-телеангиэктазией смертельно опасной. Частые заболевания легких приводят к развитию бронхоэктазов и пневмосклероза.
Злокачественные новообразования. Среди пациентов, имеющих синдром Луи-Бар, злокачественные опухолевые процессы отмечаются в 1000 раз чаще, чем в среднем у населения. Наиболее распространенными среди них являются лейкемия и лимфома.
Синонимы синдрома Луи-Бар. S. Boder—Sedgwick. Це-фало-глазо-кожная телеангиэктазия. Мозжечково-глазо-кожная телеангиэктазия. Телеангиэктатическая атаксия. Атрофия мозжечка с глазо-кожными телеангиэктазиями и бронхоэктазиями. Синдром телеангиэктазии и атаксии.
Определение синдрома Луи-Бар. Редкий факоматоз у детей. Относится к нейро-кожным синдромам.
Авторы. Louis-Bar Denise — современный французский врач. Впервые описала синдром в 1941 г. В 1957—1958 гг. Boder и Sedgwick опубликовали 8 новых наблюдений и впервые привели данные аутопсии.
Симптоматология синдрома Луи-Бар:
1. Впервые проявляющиеся в раннем детском возрасте и медленно прогрессирующие мозжечковые атаксия, абазия и астазия; ко времени полового созревания свободные походка и стояние, как правило, невозможны. Одновременно развиваются расстройства речи (монотонная скандированная речь или регулярная дизартрия), также носящие прогрессирующий характер.
2. Отсутствие пирамидных знаков, рефлексы нормальные или ослаблены. Мышечный тонус (после первичного ригидноподобного повышения) обычно снижен. Нормальная чувствительность. Отсутствие парезов.
3. Медленно развивающиеся симметричные телеангиэктазии кожи и слизистых оболочек, особенно кожи лица и конъюнктивы (ранний симптом, который может проявиться быстропроходящим конъюнктивитом!). Частое развитие бляшек цвета кофе с молоком, атрофии кожи лица, преждевременное поседение волос (в школьном возрасте).
4. Рецидивирующие легочные инфекции, иногда с развитием бронхоэктазий.
5. Гиперсаливация.
6. Малый рост и общая дистрофия.
7. К началу заболевания интеллектуальное развитие нормальное, позднее происходит задержка психического развития.
8. Пневмоэнцефалографические данные: признаки мозжечковой атрофии.
9. Атаксия — телеангиэктазия очень часто комбинируется с гипоплазией вилочковой железы, специфической дисгаммаглобулинемией (недостаточность гамма Ау, глобулина) и склонностью к злокачественным процессам в ретикулоэндотелиальной системе (лимфосаркома, ретикулезы и т. д.).
10. Прогноз плохой. Большинство наблюдавшихся до сих пор больных погибали в период полового созревания.
Этиология и патогенез синдрома Луи-Бар. Рецессивно-наследственное расстройство с генетически обусловленным торможением васкуляризации головного мозга. В одном случае была установлена транслокация между двумя акроцентрическими хромосомами группы 13—14—15 (Bijl, Jansen, Ossentjuk, 1963). До сих пор не ясно значение обнаруживаемого в отдельных случаях избыточного выделения с мочой полипептидов.
Патологическая анатомия. Первичнохроническая прогрессирующая мозжечковая дегенерация с патологическими изменениями клеток Пуркине и сморщиванием белого вещества, а также изменения вен (расширение, засстой, истончение стенок), особенно в области мягкой мозговой оболочки мозжечка, а также и больших полушарий.
Дифференциальный диагноз. В начальных стадиях: мозжечковая форма синдрома церебрального детского паралича. S. Friedreich I (см.). Опухоли мозжечка. S. Sturge—Weber (см.). S. v. Hippel—Lindau (см.). S. Werner (см.). S. Osier I (см.).
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021

1. Нестабильность генома головного мозга: этиология, патогенез и новые биологические маркеры психических болезней / А.С. Тиганов, Ю.Б. Юров, С.Г. Ворсанова, И.Ю. Юров // Вестник РАМН. – 2012. – № 9. – С. 45–53.
2. Мозаичная анеуплоидия в клетках головного мозга при атаксии-телеангиэктазии (синдроме Луи-Бар) / И.Ю. Юров, С.Г. Ворсанова, А.Д. Колотий и др. // Мед. генетика. – 2008. – № 73. – С. 22–26.
3. Генетические механизмы нейродегенерации: исследование геномной нестабильности в нервных клетках при атаксии-телеангиэктазии (синдром Луи-Бар) / И.Ю. Юров, М.К. Тагирова, А.Д. Колотий и др. // Успехи соврем. естеств. – 2012. – № 3 – С. 89–89.
4. Вариации генома соматической клетки в норме и при наследственной патологии в ходе онтогенеза / И.Ю. Юров, С.Г. Ворсанова, А.П. Сильванович, Ю.Б. Юров // Мед. генетика. – 2012. – Т.11. – № 6. – С.11–20.
5. Юров Ю.Б., Ворсанова С.Г. Молекулярно-цитогенетические исследования хромосомных аномалий и нарушений при нервно-психических заболеваниях: поиск биологических маркеров для диагностики // Вестник РАМН. – 2001. – С. 26–31.
6. Юров И.Ю., Ворсанова С.Г., Юров Ю.Б. Молекулярная нейроцитогенетика: нестабильность генома в мозге при психических заболеваниях // Психиатрия. – 2007. – Т. 28. – № 4. – С. 36–43.
7. Нестабильность хромосом в нервных клетках человека в норме и при нервно-психических заболеваниях / Ю.Б. Юров, С.Г. Ворсанова, И.Ю. Соловьев, И.Ю. Юров // Генетика. – 2010. – Т. 46. – № 10. – С. 1352–1355.
8. Юров И.Ю., Ворсанова С.Г., Юров Ю.Б. Вариабельность и нестабильность генома в клетках головного мозга при психических и нейродегенеративных заболеваниях // Психиатрия. – 2010. – № 3. – С. 7–12.
10. Aurias A., Dutrillaux B. Probable involvement of immunoglobulin superfamily genes in most recurrent chromosomal rearrangement from ataxia telangiectasia // Hum.Genet. – 1986. – № 72. – Р. 210–214.
11. Barker R.A., Cicchetti F. Current understanding of the glial response to disorders of the aging CNS // Front. Pharm. – 2012. – Vol. 3. – P. 1–5.
12. Bochkov N.P., Lopukhin Y.M., Kuleshov N.P. et al. Cytogenetic study of patients with ataxia-telangiectasia // Humangenetik. – 1974. – Р. 115–128.
13. Fischer H.G., Morawski M., Brückner M.K. еt al. Changes in neuronal DNA content variation in the human brain during aging // Aging Cell. – 2012. – Vol. 11. – № 4. – Р. 628–28.
15. Gatti R.A., Berkel I., Boder E. et al. Localization of an ataxia telangiectasia gene to chromosome 11q22-23 // Nature. – 1988. – Vol. 33. – № 6199. – Р. 577–580.
16. Iourov I.Y., Vorsanova S.G., Yurov Y.B. Chromosomal variation in mammalian neuronal cells: known facts and attractive hypotheses // Int. Rev. Cytol. – 2006. – № 249. – Р. 143–191.
17. Iourov I.Y., Vorsanova S.G., Yurov Y.B. Intercellular genomic (chromosomal) variations resulting in somatic mosaicism: mechanisms and consequences // Curr. Genomics. – 2006. – № 7. – Р. 435–446.
18. Iourov I.Y., Vorsanova, S.G., Yurov Y.B. Ataxia telangiectasia paradox can be explained by chromosome instability at the subtissue level // Med. Hypotheses. – 2007. – № 68. – Р. 716.
19. Iourov I.Yu, Vorsanova S.G., Yurov Y.B. Chromosomal mosaicism goes global // Mol. Cytogen. – 2008. – Vol.1. – № 26. – Р. 1–7.
20. Iourov I.Y., Vorsanova S.G., Yurov Y.B. Molecular cytogenetics and cytogenomics of brain diseases // Curr. Genomics. – 2008. – № 9 –Р. 452–465.
21. Iourov I.Y., Vorsanova S.G., Liehr T., Yurov Y.B. Aneuploidy in the normal, Alzheimer’s disease and ataxia-telangiectasia brain: differential expression and pathological meaning // Neurobiol. Dis. – 2009. – № 34. – Р. 212–220.
22. Iourov I.Y., Vorsanova S.G., Liehr T. Increased chromosome instability dramatically disrupts neural genome integrity and mediates cerebellar degeneration in the ataxia-telangiectasia brain // Hum. Mol. Genet. – 2009. – № 18. – Р. 2656–2669.
23. Iourov I.Y., Vorsanova S.G., Yurov Y.B. Genomic landscape of the Alzheimer’s disease brain: chromosome instability – aneuploidy, but not tetraploidy – mediates neurodegeneration // Neurodegener. Dis. – 2010. – № 8. – Р. 35–37.
24. Iourov I.Y., Vorsanova S.G., Yurov Yu.B. Somatic genome variations in health and disease // Curr. Genomics. – 2010. – Vol. 11. – № 6. – P. 387. – 396.
25. Iourov I.Y.,Vorsanova S.G.,Yurov Y.B. Single cell genomics of the brain: focus on neuronal diversity and neuropsychiatric diseases // Curr.Genomics. – 2012. – № 13. – Р. 477–488.
27. Iourov I. To see an interphase chromosome or: How a disease can be associated with specific nuclear genome organization // BioDiscovery – 2012. – № 4: 5; DOI: 10.7750/ Bio Discovery. 2012.4.5.
29. McKinnon P.J. ATM and the Molecular Pathogenesis of Ataxia Teleagiectasia//The Ann. Rev. Pathol. Mechan. Dis. – 2012. – № 7. – P. 303–321.
30. Mezey E., Chandross K.J., Harta G. et al. Turning blood into brain: cells bearing neuronal antigens generated in vivo from bone marrow // Science. – 2000. – № 290. – Р. 1779–1782.
31. Mostafa M., Aghamohammadi A., Kouhi A. et al. Ataxia-telangiectasia in Iran: Clinical and features of 104 patients // Pediatr.Veurol. – 2007. – Vol. 37. – № 1. – P. 21–28.
32. Muotri A.R., Gage, F.H. Generation of neuronal variability and complexity // Nature. – 2006. – № 441. – Р. 903–910.
33. Oxford J.M., Harnden D.G., Parrington J.M. et al. Specific chromosome aberrations in ataxia telangiectasia // J.Med.Genet. – 1975. – № 12. – Р. 251–262.
34. Pandita T.K., Pathak S., Geard C.R. Chromosome end associations, telomeres and telomerase activity in ataxia-telangiectasia cells // Cytogenet.Cell Genet. – 1995. – Vol. 71, № 1. – Р. 86–93.
35. Ponti G., Peretto P., Bonfanti L. Genesis of neuronal and glial progenitors in the cerebellar cortex of peripuberal and adult rabbits // Plos ONE. – 2008. – № 3. – Р. 2366.
36. Vorsanova S.G., Yurov Y.B., Iourov I.Y. Human interphase chromosomes: a review of available molecular cytogenetic technologies // Mol. Cytogenet. – 2010. – № 3. – Р. 1.
37. Vorsanova S.G., Yurov Y.B., Soloviev I.V., Iourov I.Y. Molecular cytogenetic diagnosis and somatic genome variations // Curr. Genomics. – 2010. – Vol. 11. – № 6. – P. 440–446.
38. Yurov Y.B., Iourov I.Y., Monakhov V.V. et al. The variation of aneuploidy frequency in the developing and adult human brain revealed by an interphase FISH study // J.Histochem. Cytochem. – 2005. – Vol. 53. – № 1. – P. 385–390.
39. Yurov Y.B., Iourov I.Y., Vorsanova S.G. et al. The schizophrenia brain exhibits low-level aneuploidy involving chromosome 1 // Schizophr. Res. – 2008. – № 98. – Р. 139–147.
40. Yurov Y.B., Iourov I.Y., Vorsanova S.G. et al. Aneuploidy and confined chromosomal mosaicism in the developing human brain // PLoS ONE. – 2007. – № 2. – Р. 558.
41. Yurov Y.B., Vorsanova S.G., Iourov I.Y. et al. Unexplained autism is frequently associated with low-level mosaic aneuploidy // J. Med. Genet. – 2007. – № 44. – Р. 521–525.
42. Yurov Y.B., Vorsanova S.G., Iourov I.Y. GIN’n’CIN hypothesis of brain aging: deciphering the role of somatic genetic instabilities and neural aneuploidy during ontogeny // Mol. Cytogenet. – 2009. – № 2. – Р. 23.
43. Yurov Y.B., Vorsanova S.G., Iourov I.Y. Ontogenetic variation of the human genome // Curr Genomics. – 2010. – № 11(6). –P. 420–425.
44. Yurov Y.B., Vorsanova S.G., Iourov I.Y. The DNA replication stress hypothesis of Alzheimer’s disease // ScientificWorldJournal. – 2011. – № 11. – Р. 2602–2612.
Нестабильность генома на хромосомном уровне (численные и структурные аномалии хромосом) является наиболее частой генетической причиной нарушения центральной нервной системы (ЦНС). В частности, по данным разных исследователей, подобные формы геномных вариаций связаны с 30–50 % случаев умственной отсталости у детей, 5–40 % – аутизма и 1–10 % – шизофрении [1, 4, 5, 16]. Известно также, что патогенез нейродегенеративных заболеваний, например, болезни Альцгеймера, связан с субмикроскопическими вариациями на хромосомном уровне, а также анеуплоидизацией тканей ЦНС [1, 6–8, 23, 44]. Более того, некоторые заболевания, которые рассматривают также в качестве молекулярных моделей нейродегенерации, представляют собой синдромы хромосомной нестабильности (например, атаксия-телеангиэктазия или АТ) [16, 28, 29]. Это позволяет сделать вывод о том, что геномные вариации и нестабильность, в частности, анеуплоидия, являются одним из значимых генетических факторов предрасположенности ко многим психическим и нейродегенеративным заболеваниям, а также связаны со старением мозга [1, 4, 17, 20, 24, 42].
Парадоксальная особенность АТ, которая отмечается многими исследователями, связана с прогрессирующей мозжечковой дегенерацией, которая при наличии мутации в гене АТМ во всех клетках головного мозга, наблюдается преимущественно в мозжечке [28, 29]. Именно прогрессирующая мозжечковая дегенерация является основной причиной нарушений функций мозжечка и связана с высокой и ранней летальностью при этом неизлечимом заболевании ЦНС. Характерно, что другие отделы мозга больных не поражаются нейродегенерацией или нейродегенеративные процессы в них наступают на более поздних сроках заболевания [14, 29]. МакКиннон [28, 29], по-видимому, впервые обратил особое внимание на этот парадоксальный феномен, предположив, что ответ на вопрос о специфике нейродегенеративных процессов в разных отделах мозга при АТ позволит выяснить загадочную природу этого неизлечимого заболевания и разработать научно обоснованные методы его терапии.
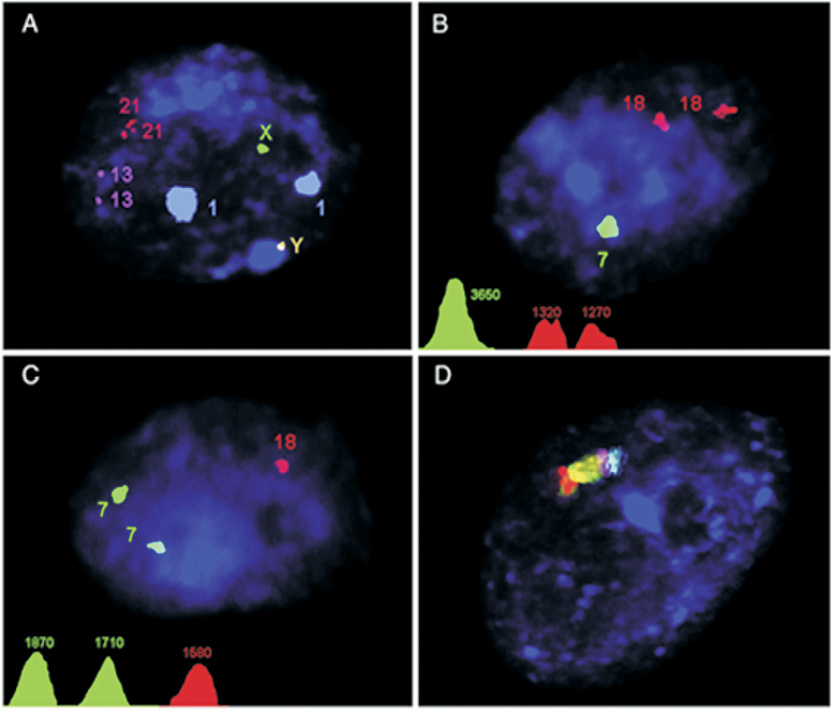
Таким образом, для объяснения парадокса МакКиннона [28, 29] и экспериментальной проверки оригинальной гипотезы Юрова И.Ю. с соавторами [18] о связи нестабильности генома с нейродегенерацией нами был проведен молекулярно-цитогенетический анализ особенностей CIN в клетках головного мозга больных с АТ (аутопсийные образца коры и мозжечка). Исследование хромосомных разрывов и анеуплоидии проведено с помощью многоцветовой интерфазной флюоресцентой гибридизации in situ (MFISH), интерфазного хромосомоспецифичного многоцветового окрашивания (ICS-MCB), иммуно-FISH и биоинформатического анализа как описано ранее [2, 21–22]. Примеры молекулярно-цитогенетического анализа анеуплоидии в клетках головного мозга в норме и при АТ показаны на рис. 1, 2.
В мозге больных АТ по сравнению с контролем выявлено 2–5-кратное (до 20–50 %) увеличение уровня анеуплоидии в коре и мозжечке. Кроме того, было выявлено 5-20-кратное увеличение числа клеток со специфическими разрывами хромосомы 7, 14 и Х в нервных клетках мозжечка, в то время как в других отделах мозга хромосомные аномалии не наблюдались [22]. Таким образом, наши данные в целом согласуются с гипотезой о патогенной роли нестабильности соматического генома в клетках мозжечка при АТ, и, по-видимому, представляют собой возможное объяснение парадокса МакКиннона.
Эти исследования открыли новые особенности организации и функционирования генома в клетках головного мозга при АТ, которые не удается объяснить, оперируя имеющимися в литературе данными. Во-первых, не ясно, почему резкое возрастание спонтанного уровня анеуплоидии при АТ, затрагивающее различные отделы мозга (префронтальная кора и мозжечок), не приводит к нарушениям функционирования мозга больных, например, как это наблюдается при хромосомных болезнях. Известно, что регулярные и мозаичные формы хромосомных болезней всегда негативно затрагивают функции головного мозга и проявляются в виде различных форм умственной отсталости, аутизма и эпилепсии [5–8, 17, 20]. Парадоксально, но этих психических нарушений при АТ нет (новый парадокс 1). Большинство исследователей не фиксируют нарушений когнитивных функций или психических заболеваний при АТ, а, наоборот, отмечают наличие нормального уровня интеллекта у больных АТ [14]. Тем не менее, имеются данные о том, что такие отклонения психики как умственная отсталость, встречаются у больных с АТ в 10 раз чаще, чем в нормальной популяции [31]. Связан ли этот новый парадокс с высокой пластичностью мозга или другими компенсаторными механизмами, блокирующими негативное влияние спонтанной анеуплоидии на функционирование ЦНС, или с недостаточно подробно описанным психическим статусом больных АТ, в настоящее время неясно.
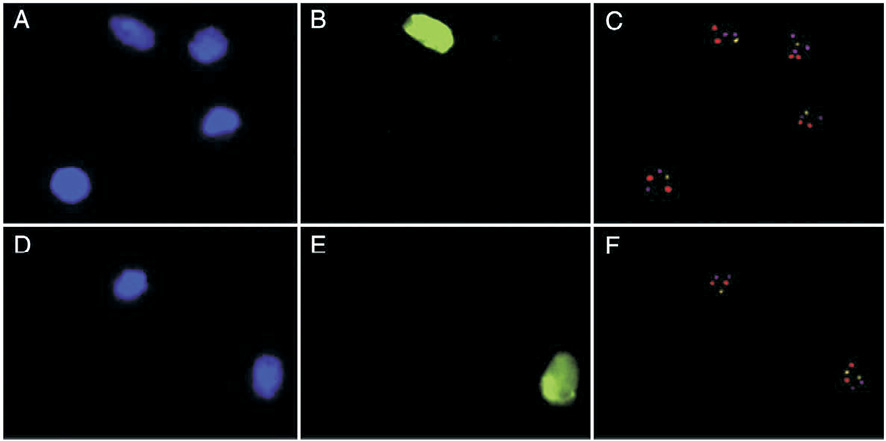
Методами молекулярной цитогенетики нами было показано, что в клетках головного мозга при АТ выявляются специфические маркеры геномной и хромосомной нестабильности, связанные с нарушением процессов репарации ДНК, сегрегации хромосом и, по-видимому, процесса запрограммированной клеточной гибели (апоптоза). Однако аномальный анеуплоидный хромосомный набор и дополнительные хромосомы с разрывами, производные хромосом 7, 14 и Х, наблюдались в 5–45 % клеток разных индивидуумов с АТ только в мозжечке. Имунно-FISH с антителами на нейрон-специфичный ядерный антиген NeuN был использован для иммунофенотипирования популяции клеток в аутопсийных образцах мозга больных АТ [21, 22]. Большинство клеток (около 80 %) с хромосомными разрывами и анеуплоидией были NeuN-негативными (рис. 3). Тем не менее, применение NeuN четко показывает, что анеуплоидия затрагивает и зрелые нейроны с аномалиями генома (до 20 % клеток с анеуплоидией и хромосомными разрывами) и глиальные клетки. Таким образом, значительное увеличение числа клеток мозжечка с анеуплоидией и хромосомными разрывами затрагивает нейроны и глию и является значимым генетическим признаком мозжечковой атаксии при АТ.
Рекуррентные точки разрыва интерфазных хромосом нервных клеток, связанные с мозжечковой нейродегенерацией при АТ, были картированы нами в участках 7p14, 14q12, Xp22.1, Xp22.3 [22]. С помощью биоинформатических технологий (анализа геномных и эпигенетических баз данных) было показано, что в участке 14q12 расположены гены-кандидаты, которые, возможно, являются мишенями CIN при АТ – FOXG1B и NOVA1. Другие точки разрыва не содержат специфических генов-мишеней, однако они расположены в участках ломкости хромосом – FRA7C, FRAXB и FRAXC (рис. 4). Следовательно, ген АТ (ATM) может регулировать стабильность сайтов ломкости. Однако для выяснения роли этих генов-кандидатов, участвующих в процессах нейрональной дисфункции и гибели в мозжечке больных АТ, необходимы дальнейшие нейробиологические и генетические исследования, которые помогут определить молекулярно-генетические механизмы мозжечковой дегенерации. Таким образом, анеуплоидия и хромосомные разрывы в дегенерирующем мозжечке затрагивают специфические хромосомы (7, 14 и Х) и, по-видимому, специфические генные локусы этих хромосом, роль которых в нейродегенеративных процессах неясна [22, 24–26].
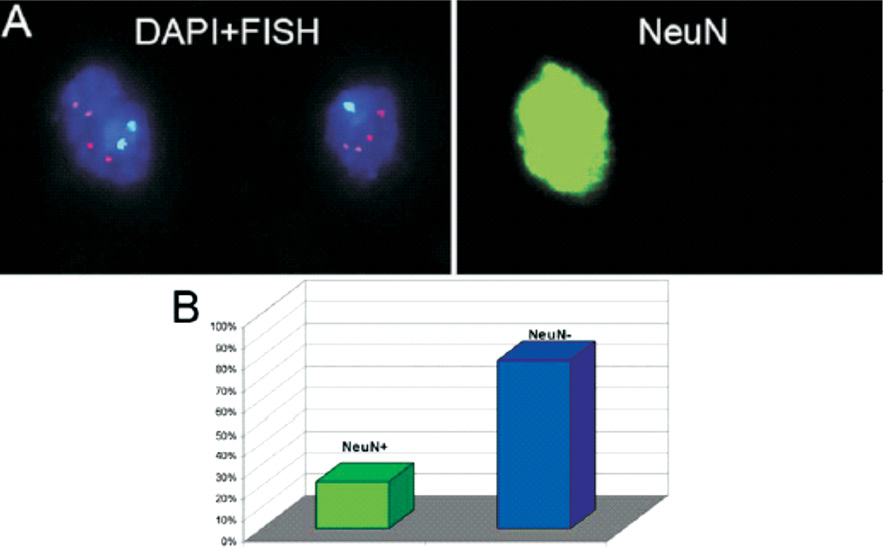
Рис. 3. Многоцветовая иммуно-FISH (NeuN иммунофенотипирование + многоцветная FISH) в клетках мозжечка при АТ (А, слева). Совместная трехцветная FISH с центромерными ДНК пробами на хромосому 1 (голубые сигналы), на 18 (розовые сигналы), и Х (красные сигналы) и окраска с DAPI, демонстрирующая одно анеуплоидное ядро (справа) (моносомия по хромосоме 1). (А, справа) NeuN иммунофенотипирование каждого ядра показывает одно NeuN- позитивное ядро нейрона (зеленый цвет, слева) с двумя хромосомами 1 и одно NeuN-негативное анеуплоидное нейронное с моносомией 1. (В) Частота NeuN-позитивных и NeuN-негативных схожих нейронов с хромосомным дисбалансом в мозжечке больных АТ. Было предположено, что генетически нестабильные NeuN-негативные нейрональные клетки могут представлять собой незрелые гранулярные нейроны –основной тип нейронов в мозжечке, а также нейрональные предшественники клеток Пуркинье, которые также являются NeuN-негативными. Кроме того, есть высокая вероятность, что они могут представлять собой также глиальные клетки, включая микроглию, которая является единственным иммунокомпетентным компартментом в ЦНС. (From [22], reproduced with permission of Springer Science + Business Media in the format reuse in a book/textbook via Copyright Clearance Center)
Хромосомоспецифические разрывы и анеуплоидия, по-видимому, представляют собой ранее неизвестную парадоксальную особенность нестабильности генома в мозге при АТ (новый парадокс 2), которая могла бы быть объяснена процессами соматической рекомбинации в нервных клетках головного мозга. Однако наличие соматической рекомбинации генома в клетках ЦНС до сих пор не доказано. Ответ на вопрос о роли специфических повреждений хромосом 14, 7 и Х при мозжечковой дегенерации требует разработки новых технологических решений, которые позволят исследовать геномные вариации в индивидуальных клетках генетически и функционально гетерогенных и мозаичных клеточных популяций головного мозга человека [25, 27].
Интересно, что при анализе спектра и частоты хромосомных аберраций у пациентов разного возраста нами обнаружена положительная корреляция частоты нервных клеток с CIN и увеличением продолжительности жизни больных АТ [22]. Эти данные не согласуются с общепринятым мнением о патогенной роли хромосомной нестабильности, так как увеличение числа аномальных анеуплоидных клеток, напротив, должно приводить к их дисфункции и ускоренной гибели, а также к тяжести клинических проявлений заболевания [13, 16, 42]. Тем не менее, у больных, доживших до старшего возраста (35–47 лет), частота CIN для хромосом 14 и 7 увеличена во много раз (от 5 до 40 %) по сравнению с более молодыми пациентами, которые умерли в возрасте 19–24 лет от осложнений при АТ (рис. 5).
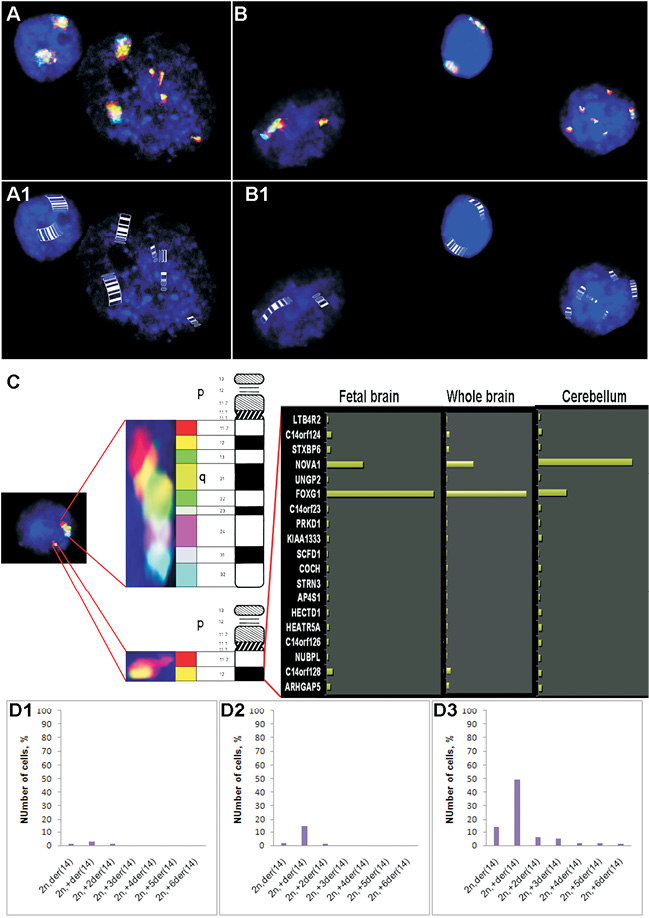
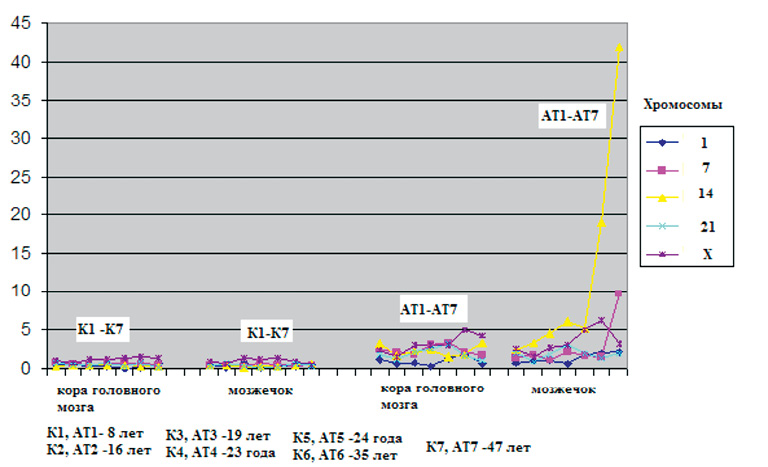
Рис. 5. Зависимость частоты анеуплоидии по хромосомам 1, 7, 14, 21, Х от возраста в диапазоне от 7 до 47 лет в группах 7 здоровых индивидуумов (К1-7) и 7 индивидуумов с АТ (АТ1-7). По оси Х – частота анеуплоидии (% %), по оси У − исследуемые аутопсийные образцы тканей головного мозга разных индивидов, ранжированные по возрасту (7, 16, 19, 23, 24, 35 и 47 лет). Видно резкое возрастание числа клеток с CIN, затрагивающих хромосомы 14 и 7 в мозжечке у больных старшего возраста
Работа выполнена в рамках плановых научных исследований НЦПЗ РАМН и частично поддержана грантом РФФИ № 12-04-00215-а.
Медико-генетический научный центр РАМН
Медико-генетический научный центр РАМН
Атаксия-телеангиэктазия с редким фенотипом и необычной родословной
Журнал: Журнал неврологии и психиатрии им. С.С. Корсакова. 2019;119(6): 101-106
Медико-генетический научный центр РАМН



Медико-генетический научный центр РАМН
Медико-генетический научный центр РАМН
Атаксия-телеангиэктазия (А-Т; син.: синдром Луи-Бар), OMIM 208900, — аутосомно-рецессивное заболевание, обусловленное мутациями в гене ATM (Ataxia-Telangiectasia Mutated). Болезнь, описанная в 1941 г. бельгийским неврологом Денизой Луи-Бар (Louis-Bar), встречается повсеместно и является одной из частых наследственных атаксий раннего возраста [1, 2]. В Международной классификации болезней 10-го пересмотра (МКБ-10) А-Т входит в группу мозжечковых атаксий с нарушенной репарацией ДНК по основной функции идентифицированного в 1995 г. гена [3].
Наблюдение
После этого семья обратилась в МГНЦ для подтверждения диагноза А-Т (консультация генетика, верификация генотипов членов семьи) и рекомендаций по планированию деторождения.
Таким образом, у сына и матери диагностирована А-T с разными фенотипами. Обоим рекомендовано наблюдение невролога, иммунолога, гематолога, онколога. Риск А-Т для будущих детей в семье — 50% (а не 25%, как у здоровых родителей-гетерозигот). При планировании деторождения семья настроена на доимплантационную (с ЭКО) ДНК-диагностику.
У 45-летней женщины, единственной больной в немецкой семье, дистония возникла в детстве на фоне задержки двигательного и речевого развития; клиническая картина включала кривошею, дистонический тремор головы, оромандибулярную дистонию, дисфагию; атаксии, телеангиэктазы, подверженности инфекциям не было. Препараты L-ДОФА, ботулотоксина, бензодиазепины, глубинная стимуляция бледного шара не дали эффекта. Методом WES были найдены мутации ATM: ранее описанная c.8147T>C и новая c.8578_8580delTCT, секвенированием по Сэнгеру доказана биаллельность. Выявлены значительное повышение АФП (235 нг/мл), снижение уровня IgA при нормальном содержании IgE и IgG, умеренная лимфопения за счет Т-лимфоцитов. Детальное соматическое обследование не выявило онкологической патологии, случаи которой были в семье [15].
Описаны и другие качественно отличающиеся фенотипы А-Т. У 4 голландских больных (братья 37, 38, 43 лет и 42-летняя женщина из другой семьи) ведущим симптомом поздней А-Т была дистальная спинальная амиотрофия с началом в 29—37 лет; на момент обследования при выраженной дистальной слабости и амиотрофии ног все ходили; атаксия и телеангиэктазы были минимальны, глазодвигательные нарушения отсутствовали, наблюдался тремор покоя; МРТ, проведенная 3 больным, у 2 была нормальной, у 1 выявила легкую атрофию мозжечка. Общими признаками были повышенный АФП и пограничные с нормой изменения иммунограммы. Выявлены биаллельные мутации: в семейном случае — миссенс-мутации с.7633T>G and с. 3136 C>T в несемейном — мутация сайта сплайсинга IVS21−1G_A и миссенс-мутация с.8147T>C [18].
Как при всех наследственных болезнях с клиническим разнообразием, обсуждается вопрос гено-фенотипических корреляций. На их наличие указывает уже тот факт, что типичные и атипичные формы А-Т не сочетаются у сибсов. В ряде исследований показано, что большинство больных с типичной тяжелой А-Т имеют мутации, ведущие к обрыву синтеза белка, тогда как легкие фенотипы связаны с миссенс-мутациями, обеспечивающими остаточную активность белка ATM [2—5] В группах 14 взрослых больных (21—36 лет на момент обследования) и 53 детей с типичной А-Т доля случаев с хотя бы одной миссенс-мутацией в генотипе составила 79 и 36% соответственно [5]. Однако есть случаи очень мягких фенотипов с полным отсутствием белка [9, 16].
Как показывают описания в литературе и наше наблюдение, диагноз А-T следует иметь в виду не только у детей, но и у взрослых больных, и не только при атаксии, но при различных двигательных нарушениях неясной природы, даже легких. Диагностическую подсказку может дать уровень АФП, обычно в той или иной степени повышенный. Иногда отправной точкой для диагноза бывают соматические проявления. Сочетанная симптоматика обусловливает участие в диагностике А-T и ведении больных разных специалистов: неврологов, генетиков, педиатров, инфекционистов, иммунологов, онкологов. Выявление атипичных форм важно с точки зрения повышенного онкологического риска (хотя он ниже, чем при типичной А-Т) и прогноза потомства. Если бы спастическая кривошея у матери С. не была заметна врачу даже без целенаправленного осмотра, ее болезнь могла быть не диагностирована. Тем самым генетический риск был бы двукратно заниженным, а результат дородовой/доимплантационной ДНК-диагностики — на 50% ложноотрицательным.
Работа выполнена в рамках государственного задания Минобрнауки России.
Читайте также: