
Синдром ди джорджи реферат
Обновлено: 02.07.2024
Введение
Глава 1. Современные методы и проблемы диагностики наследственной патологии
1.1 Современные представления о наследственных заболеваниях
1.2 Геномный импринтинг
1.3 Болезни импринтинга4
Глава 2. Характеристика синдромов Прадера-Вилли и Ангельмана
2.1 Цитогенетическая характеристика синдромов Прадера-Вилли и Ангельмана
2.2 Клинические проявления и методы диагностики синдромов Прадера-Вилли и Ангельмана
2.2.1 Клинические проявления синдрома Прадера-Вилли
2.2.2 Клинические проявления синдрома Ангельмана
2.3 Связь синдромов Прадера-Вилли и Ангельмана
Глава 3. Синдром Ди Джорджи (Ди Георга)
Выводы
Список использованной литературы
Работа содержит 1 файл
курсовая.docx
Глава 1. Современные методы и проблемы диагностики наследственной патологии
1.1 Современные представления о наследственных заболеваниях
1.2 Геномный импринтинг
1.3 Болезни импринтинга4
Глава 2. Характеристика синдромов Прадера-Вилли и Ангельмана
2.1 Цитогенетическая характеристика синдромов Прадера-Вилли и Ангельмана
2.2 Клинические проявления и методы диагностики синдромов Прадера-Вилли и Ангельмана
2.2.1 Клинические проявления синдрома Прадера-Вилли
2.2.2 Клинические проявления синдрома Ангельмана
2.3 Связь синдромов Прадера-Вилли и Ангельмана
Глава 3. Синдром Ди Джорджи (Ди Георга)
Список использованной литературы
Одной из наиболее актуальных проблем современной медицинской генетики является определение этиологии и патогенеза наследственных заболеваний. Цитогенетические и молекулярные исследования имеют высокую диагностическую информативность и ценность при решении данной проблемы, так как хромосомные аномалии встречаются с частотой от 4 до 34% при различных наследственных синдромах. В последние годы появились новые методы цитогенетических и молекулярно-цитогенетических исследований, значительно расширяющие диагностические возможности при заболеваниях, сопровождающихся различными хромосомными нарушениями. Данный обзор посвящен вопросам выбора необходимых цитогенетических или молекулярно-генетических анализов при различных формах генетических синдромов.
В работе рассматриваются генетические заболевания, вызванные микроделециями хромосом. На примере наследственных заболеваний (синдромах Прадера-Вилли, Ангельмана, Ди Джорджи) будет изучено явление геномного импринтинга, влияние которого ученые изучают и сейчас, так как до конца механизм геномного импринтинга пока не известен. Будут рассмотрены возможные варианты медицинской помощи при данных заболеваниях и риски дальнейших проявлений этих заболеваний в семьях, где встречаются такие патологии. Изучение этой проблемы на сегодня является актуальной.
Целью работы является: изучение цитогенетических и клинических проявлений микроделяционных синдромов Прадера-Вилли, Ангельмана и Ди Джорджи.
ГЛАВА 1. СОВРЕМЕННЫЕ МЕТОДЫ И ПРОБЛЕМЫ ДИАГНОСТИКИ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
1.1 Современные представления о наследственных заболеваниях
Одной из наиболее актуальных проблем современного здравоохранения является организация медико- генетической помощи семьям, где встречаются случаи рождения детей с наследственными заболеваниями. Актуальность этой проблемы определяется широким распространением данной патологии, трудностью дифференциальной диагностики большого количества наследственных болезней, проявляющихся множественными аномалиями развития с высоким риском повторения заболевания в семье.
В настоящий момент появились новые методы цитогенетического и молекулярно- цитогенетического исследований, значительно расширяющие спектр современных диагностических возможностей. К этим методам можно отнести FISH-метод, основанный на гибридизации флюоресцентно меченных ДНК-зондов на разные участки генома; сравнительную геномную гибридизацию; метод спектрального кариотипирования; праймерное исследование in situ и др. В связи с разнообразием современных цитогенетических методов в литературе широко обсуждается вопрос о выборе необходимого цитогенетического или молекулярно-генетического анализа для установления диагноза при задержках психического развития и умственной отсталости [4].
В соответствии с рекомендациями согласительной конференции американской коллегии медицинской генетики (The Consensus Conference of the American College of Medical Genetics, 1997), любому пациенту с такой патологией на первом этапе необходимо провести стандартный цитогенетический анализ c разрешением в 500 сегментов. В случае подозрения на синдром, обусловленный микроструктурными аномалиями, необходимо проведение более чувствительных молекулярно-цитогенетических исследований. Первые микроструктурные нарушения хромосом, ассоциированные с определенным синдромом, были обнаружены в 1980 г. Е. Buhler и соавторами. Они сообщили о терминальной делеции сегмента 8q24 у девочки с клиническими признаками синдрома Лангера-Гидеона. К настоящему времени насчитывается около двух десятков микроделеционных синдромов, обусловленных терминальными и интерстициальными делециями разных хромосом (Рубинштейна-Тейби, Миллера-Дикера, Вильямса, Лангера-Гидеона, Прадера-Вилли, Ангельмана, Аладжилла, Ди Джорджи, Рассела-Сильвера и др.) [5, 9-12].
- субмикроскопическая терминальная делеция 8pter, связанная с транслокацией t(8;20), приводящая к психическим и поведенческим проблемам;
- терминальная делеция хромосомы 5р у пациента с фенотипическими проявлениями синдрома Lujan-Fryns;
- тандемная транслокация 22/15 с делецией района 22q13.3 и сохранением района ядрышкового организатора хромосомы у пациента с задержкой психомоторного и речевого развития и гипотонией, без каких-либо дисморфологических особенностей;
- делеция района 22q13 у пациентов с задержкой психомоторного развития и речи, гипотонией и незначительными малыми аномалиями;
- делеция 16р, возникшая de novo, сочетающаяся с гипотонией и неспецифическими аномалиями; - субтеломерная делеция вследствие семейной сбалансированной транслокации t(3;16) (q29; р13.3), сегрегирующей в двух поколениях;
- делеция района 1р36.3 (выполнен комплексный геномный анализ карт сцепления) [2, 4]. Данная хромосомная аномалия может быть связана с гипотонией, аномалиями роста, характерным лицевым фенотипом (выпуклый лоб, глубоко посаженные глаза, плоская переносица, гипоплазия средней трети лица и выступающий подбородок), кардиомиопатией, расширением желудочков мозга, гипоплазией мозолистого тела, лейкодистрофией, психическими расстройствами [4].
Таким образом, для выявления генетических патологий, в настоящее время, стали широко использовать не только цитогенетические, но и молекулярно-цитогенетические методы исследований. Это позволяет более полно изучить проблемы наследственных заболеваний.
1.2 Геномный импринтинг
В середине XIX в. Грегор Мендель в своих опытах по скрещиванию гороха сделал наблюдение, которое впоследствии стало настоящей аксиомой для генетиков. Он обнаружил, что, если скрестить гомозиготное растение, имеющее гладкие семена, и гомозиготное растение с морщинистыми семенами, в потомстве все растения будут одинаковыми и дадут только гладкие семена. Этот результат не зависел от того, у какого из растении, взятых для скрещивания, - мужского или женского - семена были гладкими. Так Мендель открыл принцип эквивалентности реципрокных скрещиваний: у потомства ген действует одинаково независимо от того, от кого из родителей он унаследован.
Трудно переоценить значение этого наблюдения Менделя в истории и практике генетики. Было установлено, что такой закономерности подчиняется большое число наследственных признаков - не только у гороха, но и у многих других организмов [3].
Исключения из правила идентичности гибридов при реципрокных скрещиваниях (т. е. скрещиваниях между двумя формами, когда каждая из них в одном случае берется в качестве матери, а в другом - в качестве отца) в действительности известны давно, однако, как правило, их можно было отнести к одному из двух классов. Первый составляют признаки, которые определяются генами, расположенными в половых хромосомах: у самок млекопитающих в ядрах клеток имеется по две Х-хромосомы, у самцов - по одной Х- и одной У-хромосоме. Например, цветовая слепота и гемофилия связаны с генами X-хромосомы. Наследование этих сцепленных с полом признаков подчиняется вполне определенным правилам, согласно которым гибриды в реципрокных скрещиваниях не обязательно идентичны. Рассмотрим пример: у отца-дальтоника и нормальной по этому признаку матери ни один из сыновей не будет дальтоником. Если мать страдает дальтонизмом, а отец нет, то все сыновья окажутся дальтониками. В обоих случаях дочери будут нести ген, обусловливающий дальтонизм, но иметь нормальное зрение. Наследование и проявление признаков, сцепленных с полом, зависят от пола потомка, но не связаны непосредственно с полом того родителя, от которого унаследован признак.
Второй класс неэквивалентных реципрокных скрещиваний включает признаки, определяемые внеядерными генами. Некоторые клеточные органеллы - митохондрии в клетках животных, митохондрии и хлоропласты в клетках растений - обладают своей собственной генетической информацией. Эти органеллы передаются из поколения в поколение с цитоплазмой яйцеклеток и поэтому наследуются исключительно по материнской линии. Таков характер наследования цвета листьев у некоторых растений, а также заболевания человека, известного под названием митохондриальной энцефаломиопатии. При митохондриальном наследовании зигота, образующаяся в результате слияния половых клеток, получает митохондрии и содержащуюся в них мтДНК только через яйцеклетку [3].
Заболевание впервые описал американский педиатр и эндокринолог Анджело Мария Ди Джорджи в 1965 г. Синдром Ди Джорджа (синдром Ди Джорджи, синдром дисэмбриогенеза 3-4 жаберной дуги, врождённая аплазия тимуса и паращитовидных желёз, синдром 22q11.2) — разновидность идиопатического изолированного гипопаратиреоза; редкое врождённое заболевание с аутосомно-доминантным типом наследования. Заболевание развивается в результате нарушения эмбриогенеза 3-4 жаберных карманов, в результате которого нарушается закладка паращитовидных желёз и тимуса. Ген, непосредственно отвечающий за развитие данного синдрома, не известен. Характеризуется агенезией или дисгенезом паращитовидных (околощитовидных) желёз, аплазией тимуса (вилочковой железы), приводящей к резкому снижению популяци Т-лимфоцитов и иммунологической недостаточности, врождёнными аномалиями крупных сосудов (дефекты аорты, тетрада Фалло). Заболевание выявляют с частотой 1:4000-1:6000 в равной пропорции у мальчиков и девочек.
Клинические проявления
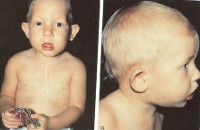
Клинически наиболее постоянными проявлениями заболевания является гипопаратиреоз и кандидомикоз, отмечаются аномалии развития носа, рта, ушей. Симптомы синдрома Ди Джорджи обусловлены нарушенными механизмами развития органов в период гестации – обычно до десятой недели беременности. Самыми яркими признаками наличия синдрома Ди Джорджи являются аномалии строения лица и ушей, недоразвитие тимуса (вилочковой железы) и паращитовидных желез, а также различные пороки сердца. Все эти патологии могут наблюдаться в различных комбинациях и проявляются такими симптомами как деформация глаз - измененный разрез, большое расстояние между глазами из-за широкой переносицы, гипертелоризм – широкая переносица, расщепление неба – волчья пасть, щели в носу, увеличение расстояния между зрачками, низко посаженные уши, симптомами гипотиреоза и сердечной недостаточности ввиду наличия дефектов сердца.
Заболевание характеризуется аплазией тимуса и связано с нарушениями развития тимуса в эмбриональном периоде. Тимусный эпителий не может обеспечить нормальное развитие Т-клеток. Отсутствие или недоразвитие тимуса является причиной иммунодефицита с преимущественным поражением его клеточного звена (Т-лимфоциты) и сохранением функций гуморального иммунитета, что в отличие от комбинированных видов иммунной недостаточности (алимфоцитоз Незелофа) не дает явных и сильно выраженных симптомов недостаточной функции иммунной системы. Иммунный статус таких детей характеризуется снижением содержания в периферической крови Т-клеток (CD3+,CD4+,CD8+) и их функциональной активности, отсутствием кожных реакций ГЗТ. Количественные показатели Т-клеток варьируют от нормы до глубокой депрессии. Характерна диссоциация между сниженными уровнями Т- и NK-клеток и повышенным содержанием В-лимфоцитов. Характерны также нормальные или повышенные уровни антител. У таких детей не отторгаются кожные трансплантаты. Изменения в гуморальном звене иммунитета не всегда развиваются, показатели гуморального иммунитета у больных с синдромом Ди Джорджа чаще всего полностью нормальны. Концентрация иммуноглобулинов всех классов нормальная. У некоторых больных повышена концентрация lgE, что, возможно, связано с отсутствием Т-супрессоров. Чаще определяется нормальное или повышенное содержание В-лимфоцитов в крови. Концентрация иммуноглобулинов в сыворотке в пределах нормы. Это свидетельствует в пользу того, что такие больные могут быть иммунизированы против ряда бактериальных и вирусных инфекций (дифтерии, столбняка, полиомиелита, кори, краснухи и др.). Так, при поражении обоих звеньев иммунитета – и клеточного, и гуморального – в большинстве случаев причиной смерти больных являются тяжелые рецидивирующие инфекции, такие как бактериальная пневмония или грибковое поражение легких, гнойные инфекции кожи и сепсис. Дети с синдромом Ди Джорджи проявляют повышенную чувствительность к вирусным, грибковым и некоторым бактериальным инфекциям. Характер течения инфекции зависит от степени недоразвития тимуса (его полной аплазии или гипоплазии). При отсутствии тимуса развиваются плохо контролируемые генерализованные инфекции, способные привести к гибели. Большую роль в исходе заболевания играет сопутствующая патология.
Врожденные пороки сердца и аномалии дуги аорты являются причиной проявления таких признаков как цианоз – синюшность кожи и слизистых оболочек, слабость и головокружение по причине гипоксии (кислородной недостаточности) тканей. Дети с некорректированными врожденными пороками сердца отстают в росте и развитии, причем отставание в умственном развитии отмечается у подавляющего большинства больных с синдромом Ди Джорджи.
Паращитовидные железы участвуют в регуляции уровня кальция в крови. Выраженная гипоплазия паращитовидных желез может стать причиной развития критического состояния, вызванного гипокальциемией. Отсутствие данных желез приводит к судорогам. Появление судорог у младенца в течение 48 часов после рождения свидетельствует о том, что у ребенка врожденная аплазия тимуса и дефицит Т-клеток. Этиологическим фактором является сниженная активность паращитовидных желез, которые в норме регулируют уровень кальция в крови, не допуская сильного его снижения, которое приводит к нарушению функций нервной системы и других тканей организма. Возможно течение синдрома в виде изолированной недостаточности паращитовидных желёз или врождённого отсутствия околощитовидных (паращитовидных) желез — гипокальциемические судороги, начиная с периода новорожденности (тетания) и вилочковой железы (различные инфекционные заболевания как следствие иммунологической недостаточности).
Одно время считали, что этот синдром вызван нарушением нормального эмбрионального развития, которое не обусловлено наследственностью, поскольку не наблюдается семейной предрасположенности к заболеванию и оба пола поражаются одинаково. Впоследствии, однако, было установлено, что многие случаи заболевания связаны со структурными дефектами в 22-й хромосоме. В целом, состояние ребенка может быть от критического до удовлетворительного.
Синдром Ди Джорджи – генетическое заболевание, относящееся к группе первичных иммунодефицитов и, наряду с ослаблением иммунитета, характеризующееся многочисленными пороками развития. Симптомами этого состояния являются частые бактериальные инфекции со склонностью к тяжелому течению, врожденные пороки сердца, аномалии развития лица и другие нарушения. Диагностика синдрома Ди Джорджи основывается на исследовании сердца, щитовидных и паращитовидных желез, изучении иммунологического статуса и данных молекулярно-генетических анализов. Лечение только симптоматическое, включает хирургическую коррекцию пороков сердца и аномалий лица, заместительную иммунологическую терапию, борьбу с бактериальными и грибковыми инфекциями.
Общие сведения
Причины синдрома Ди Джорджи
В большинстве случаев синдрома Ди Джорджи делеция 22-й хромосомы захватывает порядка 2-3 миллионов пар оснований. Чаще всего данный генетический дефект возникает спонтанно во время формирования мужских или женских половых клеток – то есть, носит герминативный характер. Лишь десятая часть всех случаев заболевания представляет собой семейную форму с аутосомно-доминантным характером наследования. Патогенез синдрома Ди Джорджи сводится к нарушению формирования особых эмбриональных образований – фарингеальных мешков (главным образом, 3-го и 4-го), которые являются предшественниками ряда тканей и органов. Главным образом, они отвечают за формирование неба, паращитовидных желез, тимуса, сосудов средостения и сердца, поэтому при синдроме Ди Джорджи возникают пороки развития именно этих органов.
Симптомы синдрома Ди Джорджи
В первые месяцы жизни больного синдромом Ди Джорджи на первый план выступают проявления врожденных пороков сердца – это может быть как тетрада Фалло, так и отдельные нарушения: дефект межжелудочковой перегородки, незаращение артериального протока и ряд других. Они сопровождаются цианозом, сердечно-сосудистой недостаточностью и при отсутствии квалифицированной медицинской помощи (в том числе и хирургической) могут приводить к ранней смерти больных. Другим распространенным нарушением у детей с синдромом Ди Джорджи считаются судороги и тетания, обусловленная гипоплазией паращитовидных желез и последующей гипокальциемией.
Следующим важнейшим проявлением синдрома Ди Джорджи, отличающим его от других разновидностей велокардиофациального синдрома, является выраженный первичный иммунодефицит. Он развивается по причине аплазии или недоразвития тимуса и поэтому в большей степени затрагивает клеточный иммунитет. Однако из-за тесных взаимосвязей между гуморальным и клеточным отделами иммунной системы это приводит к общему ослаблению защитных сил организма. Больные с синдромом Ди Джорджи крайне чувствительны к вирусным, грибковым и бактериальным инфекциям, которые нередко принимают затяжное и тяжелое течение. Некоторые исследователи отмечают наличие умственной отсталости различной степени, иногда могут наблюдаться судороги неврологического происхождения.
Диагностика синдрома Ди Джорджи
Для определения синдрома Ди Джорджи применяют метод физикального общего осмотра, кардиологические исследования (ЭхоКГ, электрокардиограмма), УЗИ щитовидной железы и тимуса, иммунологические пробы. Вспомогательную роль играет проведение общего и биохимического анализов крови, изучение анамнеза больного, генетические исследования. При осмотре больных синдромом Ди Джорджи могут определяться характерные для заболевания нарушения – расщепление твердого неба, аномалии строения лица, патологии ЛОР-органов. В анамнезе, как правило, выявляются частые эпизоды вирусных и грибковых инфекций, принимающих тяжелое течение, судороги, обусловленные гипокальциемией, нередко обнаруживается обширное кариозное поражение зубов.
На ультразвуковых исследованиях вилочковой железы отмечается значительное уменьшение массы или даже полное отсутствие органа (агенезия). ЭхоКГ и другие кардиологические методы диагностики выявляют многочисленные пороки сердца (например, дефект межжелудочковой перегородки) и сосудов средостения. Иммунологические исследования подтверждают значительное падение уровня Т-лимфоцитов. Это же явление наблюдается в периферической крови и нередко сочетается с уменьшением концентрации белков-иммуноглобулинов. Биохимическое изучение крови свидетельствует о снижении уровня кальция и гормонов паращитовидной железы. Врач-генетик может выполнить поиск делеций в 22-й хромосоме посредством флуоресцентной гибридизации ДНК или мультиплексной полимеразной цепной реакции.
Лечение синдрома Ди Джорджи
Специфического лечения синдрома Ди Джорджи на сегодняшний момент не существует, используют только паллиативные и симптоматические методики. Очень важно как можно раньше выявить врожденные пороки сердца и при необходимости произвести их хирургическую коррекцию, поскольку именно сердечно-сосудистые нарушения являются наиболее частой причиной неонатальной смерти при этом заболевании. Значительную опасность представляют собой судорожные приступы, обусловленные гипокальциемией, что требует своевременной коррекции электролитного баланса плазмы крови. Помощь хирургов при синдроме Ди Джорджи также может потребоваться для устранения пороков развития лица и неба.
Из-за выраженного иммунодефицита любые признаки бактериальной, вирусной или грибковой инфекции являются поводом для срочного применения соответствующих препаратов (антибиотиков, противовирусных и фунгицидных средств). Для улучшения иммунного статуса больного синдромом Ди Джорджи может производиться заместительное вливание иммуноглобулинов, полученных из донорской плазмы. В отдельных случаях осуществлялась пересадка вилочковой железы, которая стимулировала образование собственных Т-лимфоцитов – это способствовало улучшению качества жизни больных.
Прогноз и профилактика синдрома Ди Джорджи
Прогноз синдрома Ди Джорджи большинством исследователей оценивается как неопределенный, так как данное заболевание характеризуется значительной вариабельностью симптомов. В тяжелых случаях имеется высокий риск ранней неонатальной смерти из-за сочетания сердечно-сосудистых и иммунологических нарушений. Более доброкачественные формы синдрома Ди Джорджи требуют достаточно интенсивной паллиативной терапии, особенно важно уделять внимание лечению и профилактике вирусных и грибковых инфекций. Интеллектуальное развитие больных несколько замедлено, однако при правильной педагогической и психологической коррекции проявления задержки развития можно нивелировать. Из-за частого спонтанного характера мутаций профилактика синдрома Ди Джорджи не разработана.
Синдром Ди Джорджи (вело-кардио-фациальный синдром, синдром делеции 22q11) связан с дефектом развития четвертого глоточного кармана и четвертой жаберной дуги. Данный синдром имеет генетически неоднородный характер с частичной моносомией проксимального плеча 22-й хромосомы (при микроскопическом исследовании выявляется в трети случаев) и высокой частотой делеций (88%), выявленных по результатам FISH-исследований (Driscoll et al., 1993). В редких случаях заболевание сочеталось делецией 17р и 10p (Levy-Mozziconacci et al., 1994).
Уровень распространения синдрома делеции 22q11 (22Q11DS оценивается приблизительно как 3 случая на 10000 новорожденных (Oskarsdottir 2005) и возможно несколько чаще встречается среди девочек (личные неопубликованные данные).
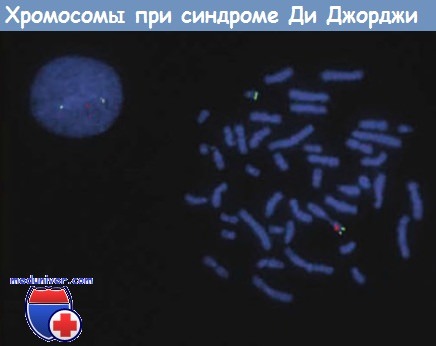
Анализ методом FISH обеих метафазных хромосом и интерфазной клетки пациента с синдромом Ди Джорджи,
демонстрирующий делецию зонда TUPLE1 (официальное название — HIRA), который локализуется на хромосоме 22q11.2.
Красным светится зонд TUPLE1, зеленым — контрольный зонд, локализующийся на 22q. Метафазная пластинка содержит одну 22-ю хромосому с зеленым и красным сигналами.
Другая 22-я хромосома содержит только зеленый сигнал, красный сигнал отсутствует в связи с делецией на этой хромосоме.
На интерфазной клетке также определяются две области гибридизации: зеленая и красная, что иллюстрирует делецию на хромосоме 22q11.2.
а) Клинические проявления. Тимус и паратиреоидные железы отсутствуют или эктопированы, также часто отмечается мальформация крупных сосудов основания сердца. Отсутствие тимуса может сопровождаться тяжелым иммунодефицитом, а отсутствие паращитовидных желез—тяжелыми гипокальциемическими судорогами в период новорожденности и позже. Гипокальциемия связана с паратиреоидным гормоном. Часто встречается расщелина неба, микрогнатия, низко расположенные уши и гипертелоризм (Conley et al., 1979).
Частично совпадающий фенотип, известный как вело-кардио-фациальный синдром или синдром Шпринтцена, проявляется сходными аномалиями сердца, дисморфизмом лица, расщелиной неба и задержкой умственного развития, иногда сопровождается поздним развитием психиатрических отклонений и также сочетается с делецией 22q11. Также может отмечаться гипоплазия мозжечка (Devriendt et al., 1996).
Синдром делеции сегмента 22q11 проявляется достаточно типичным поведенческим фенотипом. Отмечается задержка развития разговорной речи, но некоторые вербальные навыки, в частности словарный запас, имеют тенденцию к лучшему развитию, чем функции, связанные с достижением школьного возраста. Коэффициент IQ обычно составляет около 70, у отдельных пациентов превышает 100 и (очень редко) не достигает 40. Неуклюжесть движений является типичным проявлением данного заболевания, и клинические проявления синдрома делеции сегмента 22q11 часто полностью соответствуют критериям нарушения развития координации.
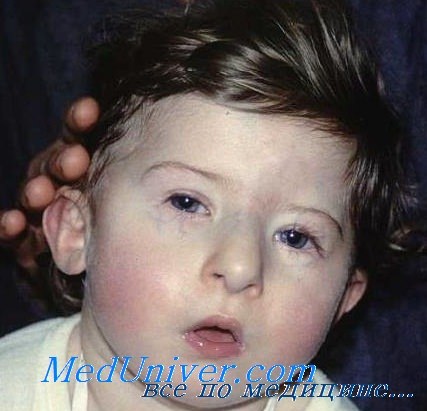
Синдром Ди Джорджи
Даже в случаях, когда признаки аутизма отсутствуют, у пациентов отмечаются социальные проблемы и проблемы общения. Данные отклонения часто сопровождаются растягиванием слов, проблемами артикуляции и бедной мимикой, характерной для синдрома делеции сегмента 22q11, так как во многих случаях отмечается небно-глоточная недостаточность. Депрессивное настроение часто отмечается, начиная с подросткового возраста. Психозы могут развиваться в позднем подростковом возрасте и раннем взрослом возрасте, даже в случаях с минимальными психиатрическими отклонениями в детском возрасте (Sobin et al., 2005). СДГВ и расстройства группы аутизма при синдроме делеции сегмента 22q11 могут быть результатом патологии головного мозга с уменьшением серого вещества коры, большого хвостатого ядра и аномалии белого вещества лобных долей и мозжечка. В подростковом и взрослом возрасте увеличивается риск развития шизофрении (Niklasson et al., 2002).
б) Лечение синдрома Ди Джорджи. Как и в случае других синдромов с поведенческим фенотипом важен ранний диагноз и обучение семьи психологической помощи. СДГВ в некоторых случаях успешно поддается лечению стимуляторами и методами когнитивно-поведенческой терапии в сочетании со специальными программами образования. Ингибиторы захвата серотонина или комбинированного захвата серотонина и норадреналина можно попробовать при длительных периодах депрессивного настроения или дистимии. Психоз (в случае его развития) может с трудом подаваться лечению, а типичные и атипичные нейролептики, несмотря на их эффективность в отношении психотических симптомов, а иногда и слуховые галлюцинации, часто менее эффективны, чем в случае других психозов у молодых людей.
Читайте также: