
Реферат на тему митохондриальные болезни
Обновлено: 05.07.2024
Митохондрии выполняют много функций, однако их основная задача — образование молекул АТФ в биохимических циклах клеточного дыхания. Основными происходящими в митохондриях процессами являются цикл трикарбоновых кислот, окисление жирных кислот, карнитиновый цикл, транспорт электронов в дыхательной цепи (с помощью I—IV ферментных комплексов) и окислительное фосфорилирование (V ферментный комплекс) [1, 2, 4, 74]. Нарушения функций митохондрий относятся к важнейшим (часто ранним) этапам повреждения клеток. Эти нарушения ведут к недостаточности энергообеспечения клеток, нарушению многих других важных обменных процессов, дальнейшему развитию клеточного повреждения вплоть до гибели клетки. Для клинициста оценка степени митохондриальной дисфункции имеет существенное значение как для формирования представлений о сути и степени происходящих на тканевом уровне процессов, так и для разработки плана терапевтической коррекции патологического состояния [6].
В меньшей степени изучены наследственные митохондриальные дефекты, связанные с повреждением ядерного генома. На сегодняшний день их известно сравнительно немного (различные формы младенческих миопатий, болезни Альперса, Лея, Барта, Менкеса, синдромы недостаточности карнитина, некоторых ферментов цикла Кребса и дыхательной цепи митохондрий). Можно предположить, что их число должно быть гораздо больше, поскольку гены, кодирующие информацию 98% митохондриальных белков, находятся именно в ядре.
На сегодняшний день существует несколько групп доказательств участия нарушения энергетических процессов в патогенезе психических заболеваний. Ниже рассматривается каждая из групп доказательств.
Нарушения психики при митохондриальных болезнях
Накопление сведений о психических расстройствах при митохондриальных болезнях стало происходить в сравнении с указанными выше нарушениями значительно позднее. Тем не менее сейчас имеется достаточное число доказательств их существования. Были описаны депрессивные и биполярные аффективные расстройства, галлюцинации и личностные изменения при синдроме Кернса—Сейра [15, 48, 71, 133], синдроме MELAS [21, 49, 66, 81, 92, 93, 95, 106, 109, 114, 122, 124], хронической прогрессирующей наружной офтальмоплегии [116] и наследственной оптической нейропатии Лебера [88, 126].
Общие признаки, характерные для митохондриальных и психических заболеваний
Речь идет об определенном клиническом сходстве некоторых психических заболеваний и митохондриальных синдромов, а также общих типах их наследования.
Прежде всего обращают на себя внимание данные о превалировании случаев наследования по материнской линии некоторых психических заболеваний, в частности биполярных расстройств [65]. Такое наследование не может быть объяснено с позиций аутосомных механизмов, а равное количество мужчин и женщин среди пациентов с биполярными нарушениями делает маловероятным предположение о возможности в данном случае Х-сцепленного наследования. Наиболее адекватным объяснением при этом может быть концепция передачи наследственной информации через митДНК. Существует также тенденция к материнскому типу наследования и у больных шизофренией [45, 108, 131]. Правда, в этом отношении имеется альтернативное используемому в нашем контексте объяснение: предполагается, что данная тенденция может обусловливаться неравными условиями больных разного пола в поиске партнера [123].
В заключение укажем еще на одно сходство: определяемое с помощью магниторезонансной томографии повышение плотности белого вещества отмечается не только при биполярных аффективных нарушениях и большой депрессии с поздним дебютом [8], но и в случаях развития ишемических изменений при митохондриальных энцефалопатиях [11].
Признаки митохондриальной дисфункции при психических заболеваниях
Шизофрения
Как говорилось выше, упоминания о признаках лактатацидоза и некоторых других биохимических изменений, свидетельствующие о нарушении энергообмена при шизофрении, начали появляться с 30-х годов ХХ века. Но только начиная с 90-х годов число соответствующих работ стало нарастать особенно заметно, причем вырос и методический уровень лабораторных исследований, что нашло отражение в ряде обзорных публикаций [13, 34, 42, 52].
На основе опубликованных работ D. Ben-Shachar и D. Laifenfeld [13] разделили все признаки митохондриальных нарушений при шизофрении на три группы: 1) морфологические нарушения митохондрий; 2) признаки нарушения системы окислительного фосфорилирования; 3) нарушения экспрессии генов, ответственных за митохондриальные белки. Это деление может быть подкреплено примерами из других работ.
При аутопсии мозговой ткани больных шизофренией L. Kung и R. Roberts [69] было выявлено снижение числа митохондрий во фронтальной коре, хвостатом ядре и скорлупе. При этом было отмечено, что оно было менее выражено у больных, получавших нейролептики, в связи с чем авторы сочли возможным говорить о нормализации митохондриальных процессов в мозге под влиянием нейролептической терапии. Это дает основание упомянуть и статью Н.С. Коломеец и Н.А. Урановой [67] о гиперплазии митохондрий в пресинаптических терминалях аксонов в области substantia nigra при шизофрении.
L. Cavelier и соавт. [20], исследуя аутопсийный материал мозга больных шизофренией, выявили снижение активности IV комплекса дыхательной цепи в хвостатом ядре.
Приведенные результаты позволили выдвинуть предположение о первичной или вторичной роли митохондриальной дисфункции в патогенезе шизофрении. Однако исследованный аутопсийный материал относился к больным, получавшим лечение нейролептиками, и, естественно, митохондриальные нарушения были связаны с лекарственным воздействием. Отметим, что подобные предположения, часто небезосновательные, сопровождают всю историю обнаружения митохондриальных изменений в различных органах и системах при психических и других заболеваниях. Что касается возможного влияния собственно нейролептиков, то следует напомнить, что склонность к лактат-ацидозу у больных шизофренией обнаружена еще в 1932 г., почти за 20 лет до их появления.
Снижение активности различных компонентов дыхательной цепи было обнаружено во фронтальной и височной коре, а также базальных ганглиях мозга и иных тканевых элементах — тромбоцитах и лимфоцитах больных шизофренией. Это позволило говорить о полисистемном характере митохондриальной недостаточности [79, 129]. S. Whatlеy и соавт. [129], в частности, показали, что во фронтальной коре снижается активность IV комплекса, в височной — I, III и IV комплексов; в базальных ганглиях — I и III комплексов, никаких изменений при этом не было обнаружено в мозжечке. Следует отметить, что во всех исследованных участках активность внутримитохондриального фермента — цитратсинтазы — соответствовала контрольным значениям, что дало основание говорить о специфичности полученных результатов для шизофрении.
Дополнительно к рассмотренным исследованиям можно привести выполненную в 1999—2000 гг. работу J. Prince и соавт. [96, 97], которые исследовали активность дыхательных комплексов в разных участках мозга больных шизофренией. Эти авторы не обнаружили признаков изменения активности I комплекса, однако активность IV комплекса была снижена в хвостатом ядре. При этом последняя, так же как и активность II комплекса, была повышена в скорлупе и в прилежащем ядре. Причем повышение активности IV комплекса в скорлупе достоверно коррелировало с выраженностью эмоциональной и когнитивной дисфункции, но не со степенью моторных нарушений.
Следует отметить, что авторы большинства приведенных выше работ признаки нарушений энергообмена объясняли воздействием нейролептиков. В 2002 г. были опубликованы очень интересные в этом отношении данные A. Gardner и соавт. [38] о митохондриальных ферментах и продукции АТФ в мышечных биоптатах у больных шизофренией, лечившихся нейролептиками и не лечившихся ими. Они установили, что снижение активности митохондриальных ферментов и продукции АТФ было обнаружено у 6 из 8 не получавших нейролептики больных, а у находящихся на нейролептической терапии больных было установлено повышение продукции АТФ. Эти данные в определенной степени подтвердили сделанные ранее выводы в работе L. Kung и R. Roberts [69].
В 2002 г. были опубликованы результаты еще одной примечательной работы [30]. В ней была изучена активность I комплекса дыхательной цепи в тромбоцитах 113 больных шизофренией в сравнении с 37 здоровыми. Больные были разделены на три группы: 1-я — с острым психотическим эпизодом, 2-я — с хронической активной формой и 3-я — с резидуальной шизофренией. Результаты показали, что активность I комплекса была достоверно повышена по сравнению с контролем у больных групп 1 и 2 и снижена у больных группы 3. Более того, была выявлена достоверная корреляция между полученными биохимическими показателями и тяжестью клинических симптомов заболевания. Аналогичные изменения были получены при исследовании в этом же материале РНК и белка флавопротеиновых субъединиц I комплекса. Результаты этого исследования, таким образом, не только подтвердили высокую вероятность полисистемной митохондриальной недостаточности при шизофрении, но и позволили авторам рекомендовать соответствующие лабораторные методы для мониторинга заболевания.
Спустя 2 года в 2004 г. D. Ben-Shachar и соавт. [14] опубликовали интересные данные о влиянии на дыхательную цепь митохондрий дофамина, которому отводят существенную роль в патогенезе шизофрении [12, 37]. Было установлено, что дофамин может ингибировать активность I комплекса и продукцию АТФ. При этом активность IV и V комплексов не изменяется. Оказалось, что в отличие от дофамина норадреналин и серотонин на продукцию АТФ не влияют.
Примечателен сделанный в указанных выше работах акцент на дисфункции I комплекса дыхательной цепи митохондрий. Такого рода изменение может отражать относительно умеренные нарушения митохондриальной активности, более значимые с точки зрения функциональной регуляции энергообмена, чем грубые (близкие к летальным для клетки) падения активности цитохромоксидазы [3].
Кратко остановимся теперь на генетическом аспекте митохондриальной патологии при шизофрении.
В указанные годы была опубликована также серия работ группы R. Marchbanks и соавт. [76, 85, 129, 130], изучавших экспрессию как ядерной, так и митохондриальной РНК во фронтальной коре в случаях шизофрении. Они выявили, что все количественно увеличенные по сравнению с контролем последовательности имели отношение к митохондриальным генам. Была существенно повышена, в частности, экспрессия митохондриального гена 2-й субъединицы цитохромоксидазы. Четыре других гена имели отношение к рибосомальной РНК митохондрий.
Японские исследователи [90, 91], исследуя 300 случаев шизофрении, не нашли признаков мутации 3243AG (вызывающей нарушение в I комплексе при синдроме MELAS). Не было обнаружено повышенной мутационной частоты в митохондриальных генах 2-й субъединицы I комплекса, цитохрома b и митохондриальных рибосом при шизофрении в работе K. Gentry и V. Nimgaonkar [43].
R. Marchbanks и соавт. [77] обнаружили мутацию в 12027 паре нуклеотидов митДНК (ген 4-й субъединицы I комплекса), которая имелась у больных шизофренией мужчин и которой не было у женщин.
Японские исследователи K. Iwamoto и соавт. [50], изучая изменения в генах, ответственных за наследственную информацию для митохондриальных белков, в префронтальной коре при шизофрении в связи с лечением нейролептиками, получили доказательства в пользу лекарственного воздействия на клеточный энергообмен.
Приведенные выше результаты могут быть дополнены данными прижизненных исследований, которые были приведены в обзоре W. Kаton и соавт. [61]: при изучении с помощью магнитно-резонансной спектроскопии распределения фосфорного изотопа 31Р было выявлено снижение уровня синтеза АТФ в базальных ганглиях и височной доле головного мозга больных шизофренией.
Депрессия и биполярные аффективные расстройства
Японскими исследователями T. Kato и соавт. [52, 53] при магнитно-резонансной спектроскопии было установлено снижение внутриклеточной рН и уровня фосфокреатина в лобной доле головного мозга у больных с биполярными расстройствами, в том числе не получавших лечения. Этими же авторами [87] снижение уровня фосфокреатина в височной доле было выявлено у резистентных к литиевой терапии больных. Другие авторы [84, 125] нашли снижение уровня АТФ в лобной доле и базальных ганглиях больных с большой депрессией. Заметим, что сходные признаки наблюдались у больных некоторыми митохондриальными болезнями [99].
Что касается молекулярно-генетических данных, сразу следует отметить, что результаты ряда работ [50, 54—56, 118] свидетельствуют об отсутствии доказательств участия делеций митДНК в развитии расстройств настроения.
Ряд исследований полиморфизма митДНК, помимо самого факта различия ее гаплотипов у больных с биполярными нарушениями и обследуемых из контрольной группы, выявили некоторые мутации, характерные для первых, в частности, в позициях 5178 и 10398 — обе позиции находятся в зоне генов I комплекса [58, 60, 65].
Другие психические расстройства
Исследований, касающихся митохондриальной дисфункции при других психических расстройствах, немного. Часть из них упоминалась в предыдущих разделах обзора. Здесь же специально упомянем работу P. Filipek и соавт. [35], в которой были описаны 2 ребенка с аутизмом и мутацией в 15-й хромосоме, в участке 15q11-q13. У обоих детей выявлены умеренная моторная задержка развития, летаргия, выраженная гипотония, лактат-ацидоз, снижение активности III комплекса и митохондриальная гиперпролиферация в мышечных волокнах. Эта работа примечательна тем, что в ней впервые были описаны митохондриальные нарушения в симптомокомплексе заболевания, этиологически связанного с определенным участком генома.
Генеалогические данные, касающиеся возможной роли митохондриальных нарушений в патогенезе психических заболеваний
Выше мы уже упоминали о такой особенности ряда психических болезней, как повышенная частота случаев наследования по материнской линии, которая может косвенно указывать на участие митохондриальной патологии в их патогенезе. Однако в литературе существуют и более убедительные доказательства последнего.
В 2000 г. были опубликованы данные, полученные F. McMahon и соавт. [80], секвенировавших весь митохондриальный геном у 9 неродственных пробандов, каждый из которых происходил из большой семьи с передачей биполярных расстройств по материнской линии. Явных отличий гаплотипов по сравнению с контрольными семьями выявлено не было. Однако по некоторым позициям митДНК (709, 1888, 10398 и 10463) была обнаружена диспропорция между больными и здоровыми. При этом можно отметить совпадение данных по позиции 10398 с уже упоминавшимися данными японских авторов [57, 59], которые предположили, что 10398А-полиморфизм митДНК является фактором риска развития биполярных нарушений.
Заслуживает внимания работа B. Burnet и соавт. [19], которые в течение 12 мес проводили анонимный опрос больных с митохондриальными заболеваниями, а также членов их семей. В числе вопросов были касающиеся состояния здоровья родителей и ближайших родственников больных (по отцовской и материнской линиям). Были, таким образом, исследованы 55 семей (группа 1) с предполагаемым материнским и 111 семей (группа 2) с предполагаемым нематеринским типом наследования митохондриального заболевания. В результате у родственников пациентов по материнской линии, по сравнению с отцовской, была выявлена большая частота нескольких патологических состояний. Среди них наряду с мигренями и синдромом раздраженного кишечника была и депрессия. В группе 1 кишечные дисфункции, мигрень и депрессия наблюдались у бoльшего процента матерей из обследованных семей — 60, 54 и 51% соответственно; во 2-й группе — у 16, 26 и 12% соответственно (р 1 Следует отметить, что среди cоответствующих описаний большое место занимают случаи с выявленной мутацией митДНК 3243AG — общепризнанной причиной развития синдрома MELAS.
Содержание работы
Файлы: 1 файл
нирс по бх.docx
- КЛАССИФИКАЦИЯ МИТОХОНДРИАЛЬНЫХ БОЛЕЗНЕЙ………………4
- МЕХАНИЗМЫ ВОЗНИКНОВЕНИЯ МИТОХОНДРИАЛЬНЫХ БОЛЕЗНЕЙ.5
- ДИСФУНКЦИЯ МИТОХОНДРИЙ…………. ……………………………… ..8
3.1 Признаки, симптомы и заболевания…………………………………………8
Впервые митохондриальные болезни были описаны Люфтом (Luft) в 1962 году, когда обследовал 35-летнюю женщину с эутиреоидным состоянием, миопатией, чрезмерным потоотделением, нетолерантностью к высокой температуре, полидипсией с полиурией и базовой скоростью метаболизма 180% от нормы.
В настоящее время митохондриология выделилась в самостоятельное научное направление. Более того, открытие в последние годы ведущей роли митохондрий в чувствительности к лекарствам, их ключевой ролью в старении, апоптозе и нейродегенеративных расстройствах привело к созданию митохондриальной медицины. Важным ее разделом являются болезни, связанные с нарушением функции митохондрий, - митохондриальные цитопатии.[3]
1 КЛАССИФИКАЦИЯ МИТОХОНДРИАЛЬНЫХ БОЛЕЗНЕЙ
Классификация митохондриальных болезней базируется на двух принципах: 1) участие мутантного белка в реакциях окислительного фосфорилирования; 2) кодируется ли мутантный белок мтДНК или ядерной ДНК.
В класс I митохондриальных болезней входят первичные дефекты окислительного фосфорилирования. Подкласс 1а включает заболевания, возникающие в результате мутаций в генах мтДНК, которые кодируют субъединицы белков, участвующих в окислительном фосфорилировании, митохондриальные тРНК и рРНК.
В подклассе 1а митохондриальных болезней выделяют три группы в зависимости от природы мутаций: крупные делеции и дупликации мтДНК, точковые мутации и небольшие перестройки в генах, кодирующих белки, и небольшие мутации в генах тРНК и рРНК. Мутации, относящиеся к классу 1а, могут проявиться только в том случае, когда они имеются в 60 % и более мтДНК.
Митохондриальные болезни класса lb обусловлены мутациями в ядерных генах (более 70 %), кодирующих субъединицы белков, которые участвуют в окислительном фосфорилировании.
Митохондриальные болезни класса II вызваны мутациями ядерных генов, продукты которых импортируются митохондриями и нарушают транскрипцию, трансляцию или репликацию мтДНК, вызывают прямое повреждение мтДНК или репарацию таких повреждений, нарушают сборку субъединиц ферментов, участвующих в реакциях окислительного фосфорилирования или их импорт митохондриями.
В этот же класс попадают те митохондриальные болезни, которые вызываются эндогенными или экзогенными токсинами. Мы рассмотрим те митохондриальные болезни, которые попадают в класс 1а.
К классу I митохондриальных болезней относится атрофия дисков зрительных нервов Лебера. Заболевание проявляется острой или подострой потерей центрального зрения, обусловленной атрофией зрительных нервов. Атрофия дисков зрительных нервов Лебера может начаться как в детском, так и в пожилом возрасте.[5]
2 МЕХАНИЗМЫ ВОЗНИКНОВЕНИЯ МИТОХОНДРИАЛЬНЫХ БОЛЕЗНЕЙ
Повреждение митохондрий в основном возникает из-за воздействия реактивных форм кислорода (РФК) . В настоящее время считают, что большинство РФК образуется комплексами I и III, вероятно, вследствие высвобождения электронов под воздействием НАД-Н и ФАД-Н в ЦПЭ. Митохондрии используют приблизительно 85% кислорода, потребляемого клеткой, в процессе образования АТФ . В ходе нормального процесса ОФ от 0.4% до 4.0% всего употребляемого кислорода преобразуется в митохондриях в супероксидные радикалы (О2-) . Супероксид трансформируется до пероксида водорода (Н2О2) с помощью ферментов детоксикации- марганцевой супероксиддисмутазы (Mn-СОД) или цинк/медь- супероксиддисмутазы (Cu/Zn СОД) ,- а затем до воды с помощью глутатионпероксидазы (ГП) или пероксидредоксина III (ПР III) . Однако, если эти ферменты не способны достаточно быстро конвертировать РФК, такие как супероксид-радикал, до воды, происходит оксидативное повреждение и аккумулируется в митохондриях. Глутатион в ПР является одним из основных антиоксидантов в организме. Глутатион представляет собой трипептид, содержащий глутамин, глицин и цистеин. ГП требует селен в качестве кофактора.
Показано, сто супероксид in vitro повреждает железо-серный кластер, находящийся в в активном центре аконитазы, фертента цикла ТКК. Из-за этого железо вступает в реакцию с Н2О2 с образованием гидроксильных радикалов через реакцию Фентона (Fenton). Кроме того, оксид азота (NO) образуется в митохондриях с помощью митохондриальной синтазы оксида азота (МтСОА) , а также свободно диффундирует в митохондрии из цитозоля . NO реагирует с O2 с образованием другого радикала- пероксинитрита (ONOO-). Вместе эти два радикала и другие радикалы могут нанести существенное повреждение митохондриям и другим компонентам клетки.В митохондриях элементами, которые особенно подвержены воздействию свободных радикалов, являются липиды, белки, окислительно-восстановительные ферменты и мтДНК. Прямое повреждение митохондриальных белков снижает их аффинность к субстратам или коферментам и таким образом нарушают их функцию.[5]
Проблема осложняется тем, что если повреждение митохондрии произошло, то функция митохондрии может быть скомпрометирована увеличением потребностей клетки для процессов репарации энергии . Митохондриальная дисфункция может привести к цепному процессу, при котором митохондриальное повреждение влечет за собой дополнительное повреждение.
Комплекс I особенно чувствителен к воздействию оксида азота (NO). У животных, которым вводили природные и синтетические антагонисты комплекса I, как правило, наблюдается гибель нейронов. Нарушение функции комплекса I было ассоциировано с наследственной оптической нейропатией Лебера, болезнью Паркинсона и другими нейродегенеративными состояниями.
Гипергликемия индуцирует образование супероксида в митохондриях эндотелиальными клетками, который является важным медиатором диабетических осложнений, таких как сердечно- сосудистые заболевания. Образование супероксида в эндотелии также способствует развитию атеросклероза, гипертензии, сердечной недостаточности, старения, сепсиса, ишемически- реперфузионных повреждений и гиперхолестеринемии.
Медиаторы воспаления, такие как фактор некроза опухолей α (ФНОα) in vitro были связаны с митохондриальной дисфункцией и повышали образование ФРК. В модели застойной сердечной недостаточности добавление ФНОα к культуре кардиомиоцитов повышало образование РФК и гипертрофию миоцитов. ФНОα вызывает митохондриальную дисфункцию путем восстановления активности комплекса III в ЦПЭ, увеличивая образование РФК и повреждение мтДНК.
Дефицит питательных веществ или их избыток также может привести к митохондриальной дисфункции. Витамины, минералы и другие метаболиты работают как необходимые кофакторы для синтеза и функционирования митохондриальных ферментов и других составляющих, которые поддерживают функцию митохондрий , и диета с недостатком микрокомпонентов может ускорять старение митохондрий и способствовать нейродегенерации . Например, ферменты участвующие в цепи синтеза гемма, требуют достаточных количеств пиридоксина, железа, меди, цинка и рибофлавина . Недостаток питательных веществ, необходимых для каких- либо компонентов цикла ТКК или ЦПЭ, может привести к увеличению образования свободных радикалов и повреждению мтДНК.[2]
Хорошо известно, что недостаток питательных веществ является широко распространенной причиной патогенеза многих заболеваний и является главным предметом спора в здравоохранении. Недостаток железа главным посредником в развитии общего груза заболеваний, затрагивающих приблизительно 2 миллиарда людей, преимущественно женщин и детей. Это наиболее распространенный тип дефицита питательных веществ. Низкий статус содержания железа снижает активность митохондрий путем выключения комплекса IV и увеличения оксидативного стресса. Механизмы, лежащие в основе процесса влияния дефицита питательных веществ (и в некоторых случаях избыток, как при перегрузке железом) на возникновение, развитие и прогрессирование заболеваний, возникающих вследствие нарушения митохондриальных функций, к настоящему времени уже изучены.[1]
3 ДИСФУНКЦИЯ МИТОХОНДРИЙ
3.1 Признаки, симптомы и заболевания
Митохондриальная дисфункция вносит вклад в многочисленные патологические состояния. Некоторые митохондриальные заболевания обусловлены мутациями или делециями в митохондриальном геноме. Митохондрии делятся и пролиферируют с более высокой скоростью оборота, чем их клетки-хозяева, и их репликация находится под контролем ядерного генома. Если пороговая доля митохондрий в клетке является дефектной и если пороговая доля таких клеток в ткани имеет дефектные митохондрии, могут возникать симптомы дисфункции ткани или органа. Практически любая ткань может быть поврежденной, и может иметь место большое разнообразие симптомов, в зависимости от степени, с которой вовлечены различные ткани.
Оплодотворенная яйцеклетка может содержать как нормальные, так и генетически дефектные митохондрии. Сегрегация дефектных митохондрий в различных тканях во время деления этой яйцеклетки является стохастическим процессом, как и отношение дефектных митохондрий к нормальным митохондриям в конкретных ткани или клетке (хотя может происходить положительный или отрицательный отбор в отношении дефектных митохондриальных геномов во время оборота митохондрий в клетках). Таким образом, множество различных патологических фенотипов могут возникать из конкретной точковой мутации в митохондриальной ДНК. Наоборот, одинаковые фенотипы могут возникать из мутаций или делеций, воздействующих на различные гены в митохондриальной ДНК. Клинические симптомы в случае врожденных митохондриальных заболеваний часто проявляются в постмитотических тканях с высокими потребностями в энергии, таких как ткани головного мозга, мышц, зрительного нерва и миокарда, но вовлекаются также и другие ткани, в том числе эндокринные железы, печень, желудочно-кишечный тракт, почки и гемопоэтическая ткань, опять-таки в зависимости отчасти от сегрегации митохондрий во время развития и от динамики митохондриального оборота во времени.
Кроме врожденных нарушений, в которых участвуют наследственные, дефектные митохондрии, приобретенная митохондриальная дисфункция вносит вклад в заболевания, в частности, нейродегенеративные расстройства, связанные со старением, такие как болезни Паркинсона, Альцгеймера, Хантингтона. Частота соматических мутаций в митохондриальной ДНК растет экспоненциально с возрастом; уменьшенная активность дыхательной цепи обнаруживается повсеместно у стареющих людей. Митохондриальная дисфункция участвует также в экситотоксическом нейронном повреждении, таком как повреждение, ассоциированное с эпилептическими припадками или ишемией.
Митохондриальные заболевания включают в себя нарушения, вызываемые огромным разнообразием молекулярных повреждений или дефектов, причем фенотипическое проявление заболевания дополнительно усложняется стохастическими распределениями митохондрий в различных тканях.
В случаях, когда человек с мутацией в митохондриальном гене несет смесь нормальной и мутантной ДНК - мутации поначалу могут вообще не иметь внешних проявлений. Нормальные митохондрии до поры до времени обеспечивают клетки энергией, компенсируя недостаточность функции митохондрий с дефектами. На практике это проявляется более или менее длительным бессимптомным периодом при многих митохондриальных заболеваниях. Однако рано или поздно наступает момент, когда дефектные формы накапливаются в количестве, достаточном для проявления патологических признаков. Возраст манифестации заболевания варьирует у разных больных. Раннее начало заболевания приводит к более тяжелому течению и неутешительному прогнозу.
Митохондриальные мутации проявляются широким рядом клинических симптомов. Эти мутации способны вовлекать тРНК, рРНК или структурные гены и могут выражаться биохимически как дефекты всей электронно-транспортной цепи или как дефекты отдельных энзимов. Митохондриальные цитопатии поражают множественные органные системы, но, как указывалось, предпочтительно поражаются органы с высокой метаболической активностью - мозг и скелетные мышцы. Таким образом, скелетные мышцы являются тканью выбора для выявления митохондриальных болезней.
Характерные признаки митохондриальных цитопатий:
1) скелетные мышцы: низкая толерантность к физической нагрузке, гипотония, проксимальная миопатия, включающая фациальные и фарингеальные мышцы, офтальмопарез, птоз.
2) сердце: нарушения сердечного ритма, гипертрофическая миокардиопатия
3) центральная нервная система: атрофия зрительного нерва, пигментная ретинопатия, миоклонус, деменция, инсультоподобные эпизоды, расстройства психики.
4) периферическая нервная система: аксональная нейропатия, нарушения двигательной функции гастроинтестинального тракта.
5) эндокринная система: диабет, гипопаратиреоидизм, нарушение экзокринной функции панкреас, низкий рост.

Список сокращений.
(ATP) АТФ- аденозинтрифрсфат
(mtDNA) мтДНК- митохондриальная ДНК
(ox-phos) ОФ- окислительное фосфорилирование
(NADH) НАД-Н- восстановленный никотинамидадениндинуклеотид
(FADH) ФАД-Н- востановленный флавинадениндинуклеотид
(TCA) ТКК- трикарбоновые кислоты
(ЕТС) ЦПЭ- цепь передачи электронов
(PDH) ПДГ- пируватдегидрогеназа
(CoA) КоА- коэнзим А
(ТРР) ТПФ- тиаминпирофосфат
(ADP) АДФ- аденозиндифосфат
(ROS) РФК- реактивные формы кислорода
(MnSOD) Mn-СОД- Mn- супероксиддисмутаза
(ZnSOD) Zn-СОД- Zn-супероксиддисмутаза
(GPX) ГП- глутатионпероксидаза
(PRX) ПР- пероксидредоксин
(mtNOS) мтСОА- митохондриальная синтаза оксида азота
(TNF α) ФНОα- фактор некроза опухолей α
(NF-kappa β) ЯФ-каппа β- ядерный фактор- каппа β
(ppm) чнм- частей на миллион
(SDH) СДГ- сукцинатдегидрогеназа
(COX) ЦОК- цитохром-с-оксидаза
(IU) МЕ- международная единица (мера вещества)
Митохондрии выполняют роль электростанций в наших клетках. Они отвечают за создание энергии в форме аденозинтрифосфата (АТФ) и участвуют в путях передачи сигналов апоптоза. Имеющаяся в настоящее время теория гласит, что митохондрии являются потомками аэробных бактерий, которые колинизировали древних прокариот около 1-3 миллионов лет назад [1. Это способствовало эволюции первых эукариотических клеток, способных к аэробному дыханию, что являлось необходимой предпосылкой для эволюции многоклеточных организмов . В поддержку этой теории выступает наблюдение, что митохондрии- единственные субклеточные структуры, помимо ядра, содержащие ДНК. Однако, в отличие от ядерной ДНК, митохондриальная ДНК (мтДНК) не защищена гистонами . Ядерная ДНК накручивается на гистоны, которые физически защищают ДНК от повреждения свободными радикалами , а также необходимы для репарации двунитевых разрывов ДНК . Поскольку мтДНК не имеет структурной поддержки со стороны гистонов, она весьма восприимчива к повреждению.
Впервые митохондриальные болезни были описаны Люфтом (Luft) и сотр. в 1962 году, когда обследовали 35-летнюю женщину с эутиреоидным состоянием, миопатией, чрезмерным потоотделением, нетолерантностью к высокой температуре, полидипсией с полиурией и базовой скоростью метаболизма 180% от нормы. Пациентка страдала от разобщения процессов окислительного фосфорилирования (ОФ). ОФ является основным путем получения энергии в клетке. Энергия в форме АТФ образуется в митохондриях в процессе реакций, в которых электроны, высвобождающиеся из восстановленных субстратов никотинамидадениндинуклеотида (НАД-Н) и флавинадениндинуклеотида (ФАД-Н), поставляются на кислород по цепи респираторной протоновой(Н + ) помпы . Разобщение процессов ОФ приводит к образованию тепла без соответствующего образования АТФ, что влечет за собой дисфункцию общего состояния пациентки. В целях компенсации ее митохондрии увеличились в размере и умножились, что отчетливо наблюдалось при анализе гистологической мышечной биопсии.
С момента этого впервые задокументированного случая было отмечено, что митохондриальная дисфункция была свойственна почти всем патологических и токсикологическим состояниям . (Это отражено в таблицах 1-3). К таким состояниям относится саркопения неалкогольный стеатогепатит; приобретенные болезни, такие как диабет и атеросклероз; нейродегенеративные заболевания, такие как болезнь Паркинсона и болезнь Альцгеймера, и наследственные состояния, совместно именуемые митохондриальными цитопатиями.
Но поскольку симптомы варьируются от случая к случаю, в зависимости от возраста начала заболевания, от скорости прогрессирования, митохондральные дисфункции могут оказаться сложными для диагностирования при своем первом появлении. Согласно Кохену (BH Cohen), автору статьи от июля 2001 г в Cleveland Clinic Journal of Medicine, ранняя фаза может оказаться неострой и может не походить на известные митохондриальные заболевания. Кроме того, симптомы, такие как утомляемость, мышечные боли, затрудненное дыхание и боли в брюшной полости, могут быть ошибочно отнесены к коллагеновым сосудистым заболеваниям, синдрому хронической усталости, фибромиалгии или психосоматическим заболеваниям .
В настоящей статье исследуются структура и функции митохондрий, детали механизмов, посредством которых приобретенная митохондриальная дисфункция может повлечь широкий диапазон симптомов, а также предлагается целесообразный подход к лечению. Я думаю, что, поскольку клиницисты становятся более квалифицированными в вопросах важности роли митохондрий для состояния здоровья и болезни, мы сможем как можно раньше начать улучшать состояние пациентов.
Структура и функции митохондрий
Энергетическая потребность клетки контролируется числом митохондрий в каждой клетке. Одна соматическая клетка может содержать от 200 до 2000 митохондрий , в то время как зародышевые клетки человека, такие как сперматозоиды, содержат постоянное число в 16 митохондрий, а ооциты содержат их до 100 000 . Самое большое число митохондрий обнаружено в большинстве метаболически активных клеток, таких как клетки скелетной и сердечной мускулатуры, клетки печени и мозга. Митохондрии были найдены в каждой клетке человека, за исключением зрелых эритроцитов .
Митохондрии производят более чем 90% энергии клетки путем окислительного фосфорилирования . Образование энергии является результатом двух тесно скоординированных метаболических процессов- цикл трикарбоновых кислот (ТКК), известный также как цикл Кребса или цикл лимонной кислоты, и цепь передачи электронов (ЦПЭ). Цикл ТКК преобразует углеводы и жиры в АТФ, но основная его задача состоит в продуцировании коэнзимов НАД-Н и ФАД-Н, так что они тоже поступают в ЦПЭ.
Полный цикл ТКК представляется следующим образом: в результате катаболизма глюкозы в цитозоле получается 2 молекулы пирувата, которые проходят через двойную мембрану митохондрий и включаются в цикл ТКК. Поскольку молекулы пирувата проходят через мембраны, они встречают на своем пути два фермента- пируваткарбоксилазу и пируватдегидрогеназу (ПДГ). Хотя ПДГ упоминается как одиночный фермент, но фактически он представляет собой комплекс из трех отдельных ферментов- пируватдегидрогеназы, дигидролипоилтрансацетилазы и дигидролипоилдегидрогеназы. ПДГ- комплекс требует многообразия коферментов и субстратов для выполнения своей функции- коэнзим А (КоА), производного пантотеновой кислоты (витамин В5); НАД + , который содержит никотиновую кислоту (витамин В3); ФАД + , который содержащий рибофлавин (витамин В2); липоевую кислоту и тиаминпирофосфат (ТПФ), который, как видно из названия, содержит тиамин (витамин В1).
При избытке энергии (относительно высокой концентрации АТФ), активируется пируваткарбоксилаза и переносит молекулы пирувата в направлении гликонеогенеза. Когда потребность в энергии высока (при относительно низкой концентрации АТФ), две молекулы пирувата проходят через ПДГ- комплекс, и образуется две молекулы ацетилкоэнзима А (ацетил коА), поступающих в цикл ТКК. Существует 9 промежуточных продуктов в цикле ТКК. Чтобы пройти через этот цикл полностью, ферменты, катализирующие биотрансформацию промежуточных продуктов, требуют следующих кофакторов: цистеин, железо, никотиновая кислота, магний, марганец, тиамин, рибофлавин, пантотеновая кислота и липоидная кислота . Когда образуется 2 молекулы ацетил КоА, каждая молекула ацетил КоА образует 3 молекулы НАД-Н и 2 молекулы ФАД-Н, в общей сложности 6 НАД-Н и 4 ФАД-Н на одну молекулу пирувата. Кроме того, ацетил КоА может образовываться при окислении жирных кислот, которые затем требуют питательного вещества L- карнитина для переноса ацетил-КоА в митохондрии для включения его в цикл ТКК.
НАД-Н и ФАД-Н несут электроны в ЦПЭ, которая встроена во внутреннюю митохондриальную мембрану и состоит из серии пяти ферментативных комплексов, обозначаемых I-VI. Донорные электроны от НАД-Н и ФАД-Н проходят через комплекс ЦПЭ, передаваясь по электрохимическому градиенту и доставляясь на двухатомный кислород (О 2 ) по цепи респираторных протоновых (Н + ) помп.
Комплексы I-IV вовлекают убихинон (коэнзим Q10, сокращенно CoQ10). Комплекс I представляет собой НАД-Н- дегидрогеназу, или НАД-Н: убихиноноксидоредуктзу; комплекс II представлен сукцинатдегидрогеназой (СДГ), или сукцинат: убихиноноксидоредуктазой; компрлекс III является dc1- комплексом, или убихинон: цитохром-С- оксидоредуктазой; комплекс IV- это цитохром-С- оксидаза, или восстановленная цитохром С: кислород- оксидоредуктаза; и комплекс V представляет собой АТФ- синтазу или протонтранслоцирующую АТФ- синтазу . Комплексы I-V содержат флавины, в состав которых входит рибофлавин, железо-серосодержащие кластеры , медьсодержащие кластеры или железосодердащие части гема.
Убихинон переносит электроны от комплексов I и II на комплекс III. Цитохром С, железосодержащий гемопротеин с биядерным медьсодержащим центром , переносит электроны от комплекса III на комплекс IV. В ходе этого процесса протоны идут через помпу на внутреннюю митохондриальную мембрану в интермембранное пространство и создают движущую силу, которую использует комплекс V для фосфорилирования аденозиндифосфата (АДФ) АТФ- синтазой, образуя таким образом АТФ. Для нормального функционирования цикла ТКК и ЦПЭ требуются все питательные вещества, участвующие в образовании ферментов и всех кофакторов, необходимых для активации этих ферментов.
Механизмы возникновения повреждений митохондриальных повреждений
Повреждение митохондрий в основном возникает из-за воздействия реактивных форм кислорода (РФК) . В настоящее время считают, что большинство РФК образуется комплексами I и III, вероятно, вследствие высвобождения электронов под воздействием НАД-Н и ФАД-Н в ЦПЭ. Митохондрии используют приблизительно 85% кислорода, потребляемого клеткой, в процессе образования АТФ . В ходе нормального процесса ОФ от 0.4% до 4.0% всего употребляемого кислорода преобразуется в митохондриях в супероксидные радикалы (О 2 - ) . Супероксид трансформируется до пероксида водорода (Н 2 О 2 ) с помощью ферментов детоксикации- марганцевой супероксиддисмутазы (Mn-СОД) или цинк/медь- супероксиддисмутазы (Cu/Zn СОД) ,- а затем до воды с помощью глутатионпероксидазы (ГП) или пероксидредоксина III (ПР III) . Однако, если эти ферменты не способны достаточно быстро конвертировать РФК, такие как супероксид-радикал, до воды, происходит оксидативное повреждение и аккумулируется в митохондриях . Глутатион в ПР является одним из основных антиоксидантов в организме. Глутатион представляет собой трипептид, содержащий глутамин, глицин и цистеин. ГП требует селен в качестве кофактора.
Показано, сто супероксид in vitro повреждает железо-серный кластер, находящийся в в активном центре аконитазы, фертента цикла ТКК . Из-за этого железо вступает в реакцию с Н 2 О 2 с образованием гидроксильных радикалов через реакцию Фентона (Fenton) . Кроме того, оксид азота (NO) образуется в митохондриях с помощью митохондриальной синтазы оксида азота (МтСОА) , а также свободно диффундирует в митохондрии из цитозоля. NO реагирует с O 2 с образованием другого радикала- пероксинитрита (ONOO - ) . Вместе эти два радикала и другие радикалы могут нанести существенное повреждение митохондриям и другим компонентам клетки.
В митохондриях элементами, которые особенно подвержены воздействию свободных радикалов, являются липиды, белки, окислительно-восстановительные ферменты и мтДНК . Прямое повреждение митохондриальных белков снижает их аффинность к субстратам или коферментам и таким образом нарушают их функцию. Проблема осложняется тем, что если повреждение митохондрии произошло, то функция митохондрии может быть скомпрометирована увеличением потребностей клетки для процессов репарации энергии. Митохондриальная дисфункция может привести к цепному процессу, при котором митохондриальное повреждение влечет за собой дополнительное повреждение.
Комплекс I особенно чувствителен к воздействию оксида азота (NO). У животных, которым вводили природные и синтетические антагонисты комплекса I, как правило, наблюдается гибель нейронов . Нарушение функции комплекса I было ассоциировано с наследственной оптической нейропатией Лебера, болезнью Паркинсона и другими нейродегенеративными состояниями.
Гипергликемия индуцирует образование супероксида в митохондриях эндотелиальными клетками, который является важным медиатором диабетических осложнений, таких как сердечно- сосудистые заболевания . Образование супероксида в эндотелии также способствует развитию атеросклероза, гипертензии, сердечной недостаточности, старения, сепсиса, ишемически- реперфузионных повреждений и гиперхолестеринемии .
Медиаторы воспаления, такие как фактор некроза опухолей α (ФНОα) in vitro были связаны с митохондриальной дисфункцией и повышали образование ФРК . В модели застойной сердечной недостаточности добавление ФНОα к культуре кардиомиоцитов повышало образование РФК и гипертрофию миоцитов . ФНОα вызывает митохондриальную дисфункцию путем восстановления активности комплекса III в ЦПЭ, увеличивая образование РФК и повреждение мтДНК .
Дефицит питательных веществ или их избыток также может привести к митохондриальной дисфункции. Витамины, минералы и другие метаболиты работают как необходимые кофакторы для синтеза и функционирования митохондриальных ферментов и других составляющих, которые поддерживают функцию митохондрий (см. табл. 4), и диета с недостатком микрокомпонентов может ускорять старение митохондрий и способствовать нейродегенерации . Например, ферменты участвующие в цепи синтеза гемма, требуют достаточных количеств пиридоксина, железа, меди, цинка и рибофлавина . Недостаток питательных веществ, необходимых для каких- либо компонентов цикла ТКК или ЦПЭ, может привести к увеличению образования свободных радикалов и повреждению мтДНК.
Хорошо известно, что недостаток питательных веществ является широко распространенной причиной патогенеза многих заболеваний и является главным предметом спора в здравоохранении. Недостаток железа главным посредником в развитии общего груза заболеваний, затрагивающих приблизительно 2 миллиарда людей, преимущественно женщин и детей . Это наиболее распространенный тип дефицита питательных веществ. Низкий статус содержания железа снижает активность митохондрий путем выключения комплекса IV и увеличения оксидативного стресса . Механизмы, лежащие в основе процесса влияния дефицита питательных веществ (и в некоторых случаях избыток, как при перегрузке железом) на возникновение, развитие и прогрессирование заболеваний, возникающих вследствие нарушения митохондриальных функций, к настоящему времени уже изучены.
Тестирование и варианты лечения.
Митохондриа́льные заболева́ния — группа наследственных заболеваний, связанных с дефектами в функционировании митохондрий, приводящими к нарушениям энергетических функций в клетках эукариотов, в частности — человека.
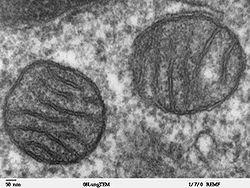
Рисунок 1 - Электронномикроскопическая фотография, показывающая митохондрии человека в поперечном сечении. Митохондриальные заболевания обусловлены генетическими, структурными, биохимическими дефектами митохондрий, приводящими к нарушениям тканевого дыхания. Они передаются только по женской линии к детям обоих полов, так как сперматозоиды передают зиготе половину ядерного генома, а яйцеклетка поставляет и вторую половину генома, и митохондрии. Патологические нарушения клеточного энергетического обмена могут проявляться в виде дефектов различных звеньев в цикле Кребса, в дыхательной цепи, процессах бета-окисления и т. д.
Не все ферменты и другие регуляторы, необходимые для эффективного функционирования митохондрий, кодируются митохондриальной ДНК. Большая часть митохондриальных функций контролируется ядерной ДНК.
Можно выделить 2 группы митохондриальных заболеваний:
Ярко выраженные наследственные синдромы, обусловленные мутациями генов, ответственных за митохондриальные белки (синдром Барта, синдром Кернса-Сейра, синдром Пирсона, синдром MELAS, синдром MERRF и другие).
2. Наследование митохондриальных болезней
Митохондрии наследуются иначе, чем ядерные гены. Ядерные гены в каждой соматической клетке обычно представлены двумя аллелями (за исключением большинства сцепленных с полом генов у гетерогаметного пола). Один аллель унаследован от отца, другой от матери. Однако митохондрии содержат собственную ДНК, причем в каждой митохондрии человека обычно содержится от 5 до 10 копий кольцевой молекулы ДНК (см. Гетероплазмия), и все они наследуются от матери. . Когда митохондрия делится, копии ДНК слеучайным образом распределяются между ее потомками, а затем происходит удвоение ДНК. Если только одна из исходных молекул ДНК содержит мутацию, в результате случайного распределения такие мутантные молекулы могут накопиться в некоторых митохондриях. Митохондриальная болезнь начинает проявляться в тот момент, когда заметное число митохондрий во многих клетках данной ткани приобретают мутантные копии ДНК (пороговая экспрессия).
Мутации в митохондриальной ДНК происходят по разным причинам намного чаще, чем в ядерной. Это означает, что митохондриальные болезни достаточно часто проявляются из-за спонтанных вновь возникающих мутаций. Иногда темп мутирования увеличивается из-за мутаций в ядерных генах. кодирующих ферменты, которые контролируют репликацию ДНК митохондрий.
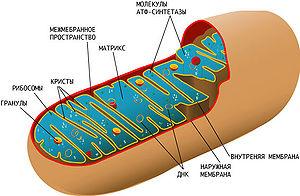
Рисунок 2 - Схема строения митохондрии. Сложная структура митохондрии и наличие собственной кольцевой хромосомы, кодирующей некоторые компоненты митохондрии, усложняет выяснение причин митохондриальных заболеваний.
Митохондриальные миопатии — это группа заболеваний мышц, которые возникают в результате нарушения функции тканевого дыхания при патологиях митохондрий. Болезни проявляются нарастающей мышечной слабостью, атрофией мускулатуры, прогрессирующими двигательными расстройствами, которые могут сопровождаться судорогами, поражениями сердца, ухудшением слуха и зрения. Диагностика предполагает лабораторные (исследование мышечных биоптатов, генетическое тестирование, биохимические анализы), инструментальные методы (ЭМГ, церебральное МР-сканирование). Лечение включает симптоматические препараты, физиотерапию, ЛФК.
МКБ-10
Общие сведения
Причины
Миопатии обусловлены наследственными либо спорадическими мутациями в митохондриальной ДНК или в ядерных генах, контролирующих работу митохондрий. Известно более 300 вариантов генных дефектов — однонуклеотидных замен, делеций, вызывающих нарушения функционирования митохондрий. Митохондриальные миопатии имеют ряд генетических аспектов, выделяющих их среди всех наследственных заболеваний. Принципиальные различия:
- Материнское наследование. Эмбрион получает всю цитоплазму с содержащимися в ней органеллами от матери, поэтому только она может передать ребенку мутантную мтДНК. В то же время, при мутациях ядерной ДНК наследование происходит по аутосомно-рецессивному, аутосомно-доминантному или Х-сцепленному типу.
- Гетероплазмия. При митохондриальной миопатии в клетках мышечной ткани одновременно присутствует мутантный и нормальный генетический материал в разном процентном соотношении, чем объясняется вариабельность клинических проявлений у членов одной семьи при наследственной форме болезни.
- Митотическая сегрегация. При делении клеток, содержащих мутантные митохондриальные гены, мтДНК распределяется между дочерними клетками случайным образом, неравномерно.
Патогенез
Митохондрия — универсальная органелла, присутствующая во всех клетках, кроме эритроцитов. Она имеет дыхательную цепь, которая включает 5 ферментных комплексов из нескольких десятков субъединиц каждый. Ферменты обеспечивают окислительное фосфорилирование для синтеза АТФ — аденозинтрифосфата, выступающего основным источником энергии в организме.
При митохондриальных заболеваниях может быть 4 варианта патогенетических механизмов развития. Как правило, при поражениях мышц наблюдаются дефекты электронного транспорта и окислительного фосфорилирования, что сопровождается нарушениями образования энергетических молекул. Другие варианты расстройств включают нарушения обмена пирувата, дефекты метаболизма жирных кислот, дисфункцию цикла Кребса.
Симптомы митохондриальной миопатии
Патологии манифестируют у детей раннего возраста, иногда они присутствуют с рождения. Общим признаком является миопатический синдром, который включает прогрессирующую мышечную слабость, снижение тонуса скелетной мускулатуры, непереносимость физических нагрузок. Типично отставание в моторном развитии: дети поздно начинают сидеть, ползать, ходить, у них сохраняется неуклюжесть движений, проблемы с поддержанием равновесия.
Клинические особенности определяются типом заболевания. При синдроме MERRF мышечная слабость сопровождается разнообразными судорожными приступами (атоническими, тонико-клоническими, миоклоническими), расстройствами координации вследствие мозжечковой атаксии. Для синдрома MELAS характерны повторные инсульты, умственная отсталость, нейросенсорная тугоухость.
У детей распространен синдром Кернса-Сайра, при котором клинические признаки дополняются расстройствами глотания, нарушениями работы проводящей системы сердца, снижением слуха. Хроническая прогрессирующая наружная офтальмопатия может возникать как компонент болезни Кернса-Сайра, так и развиваться изолированно, что чаще бывает в старшем возрасте.
Осложнения
Отличительными особенностями миопатического синдрома являются необратимость, неуклонное прогрессирование. Сначала мышечная слабость появляется в проксимальных отделах конечностей, затем поражает все тело ребенка: в процесс вовлекается гладкая мускулатура органов дыхания и пищеварения, что чревато дыхательной недостаточностью, аспирационными пневмониями, тотальным параличом. Такие пациенты теряют способность к самообслуживанию, требуют круглосуточного ухода.
Миопатии осложняются деформациями позвоночника, искривлениями нижних конечностей на фоне слабости мышечного корсета. Вследствие атрофии зрительных нервов у больных с синдромом MERRF возникает слепота. Опасным последствием многих вариантов митохондриальной патологии являются инсульты, эпилептический статус, мозговой отек, которые становятся основными причинами летального исхода.
Диагностика
Первичное обследование детей с подозрением на митохондриальную миопатию проводится у невролога, для уточнения диагноза показана консультация генетика. При осмотре учитывается неврологический статус ребенка, показатели мышечной силы и тонуса, уровень развития когнитивных навыков. Постановка диагноза требует комплексного обследования, включающего следующие методы:
Лечение митохондриальных миопатий
В клинической неврологии отсутствуют эффективные методы терапии патологии у детей. Суть медицинской помощи сводится к уменьшению моторного дефицита, своевременной коррекции осложнений, стимуляции обменных процессов в митохондриях. Наибольшую результативность демонстрируют следующие группы медикаментов:
- Аминокислоты. L-аргинин рекомендован неврологами в острой фазе для улучшения кровоснабжения мозга при осложнении миопатии инсультом, в резидуальном периоде болезни для предупреждения повторных приступов ишемии нервной ткани.
- Энерготропные препараты. Чтобы улучшить энергообеспечение тканей ребенка, эффективны препараты с левокарнитином, янтарной кислотой, коэнзимом Q10, широко используется комплекс витаминов группы В, аскорбиновая кислота, альфа-токоферол.
- Антиконвульсанты. При сочетании миопатии с эпилептическими пароксизмами показаны противосудорожные препараты группы сульфат-замещенных моносахаридов, бензодиазепинов, ограничено применяются барбитураты. Вальпроаты для лечения детей с митохондриальными нарушениями не назначаются.
Для коррекции моторных нарушений применяется расширенный комплекс физиотерапии: ультразвуковая терапия, электромиостимуляция, электрофорез с ингибиторами ацетилхолинэстеразы. Хороший эффект демонстрирует лечебный массаж, индивидуально подобранный комплекс ЛФК. Чтобы избежать перегрузки ослабленных мышц, широко используются занятия в бассейне. Также требуется ортопедическая коррекция, подбор специальной обуви ребёнку .
Прогноз и профилактика
Поскольку митохондриальные миопатии пока являются неизлечимыми болезнями, прогноз неблагоприятный. Улучшить качество жизни пациентов удается с помощью комплексной реабилитации, однако при развернутой клинической картине смерть нередко наступает в детском или молодом возрасте. Для профилактики семейным парам с отягощенной наследственностью необходимо медико-генетическое консультирование при планировании беременности.
1. Митохондриальные болезни: миопатии, энцефаломиопатии и энцефаломиелополиневропатии/ В.М. Казаков, А.А. Скоромец, Д.И. Руденко, Т.Р. Стучевская// Неврологический журнал. — 2018. — №6.
3. Влияние дисфункции митохондрий на клинические проявления наследственных миопатий/ Д.А. Харламов, В.С. Сухоруков// Российский вестник перинатологии и педиатрии. — 2013. — №4.
4. Митохондриальные миопатии в сочетании с кардиомиопатией. Новые подходы к лечению/ О.С. Страхова, Ю.М. Белозеров, С.В. Перминов, В.В. Давыдкин// Альманах клинической медицины. — 2001. — №4.
Читайте также: