
Реферат на тему миелодиспластический синдром
Обновлено: 05.07.2024
Термин "миелодиспластический синдром" был введен в 1975 г. FAB-группой, в составе которой гематологи из Франции, Америки и Великобритании, для описания ряда заболеваний, характеризующихся определенными изменениями в периферической крови, нарушением функции костного мозга и частой прогрессией в острый миелолейкоз. Данные по распространенности миелодиспластического синдрома (МДС) в различных возрастных группах представлены на рисунке.
Таблица 1. Морфологические изменения при МДС
Росток кроветворения
Костный мозг
Выделяют первичный МДС и МДС, обусловленный предшествующей терапией. В первом случае причина заболевания неясна; во втором случае возникновение МДС рассматривают как следствие цитотоксической терапии (хлорамбуцил, циклофосфамид) в анамнезе.
Таблица 2. FAB-классификация МДС
Диагностика
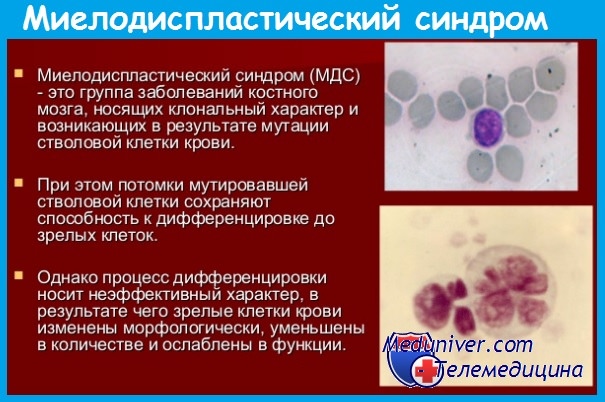
У пациентов развивается недостаточность костного мозга, которая проявляется анемией, бактериальными инфекциями, кровотечениями. У 10% больных отмечается спленомегалия. В клиническом анализе крови возможны такие изменения, как снижение гемоглобина, макроцитоз, нейтропения, моноцитоз, тромбоцитопения. Для подтверждения диагноза производят пункцию костного мозга. Изменения, выявляемые при этом, отражены в табл. 1 .
Важно помнить о том, что морфологические проявления дисплазии нельзя отождествлять с МДС, так как исходные изменения могут отмечаться при недостаточности витамина В12, фолатов, при злоупотреблении алкоголем, после цитотоксической химиотерапии, при ВИЧ-инфекции. В небольшом количестве измененные клетки встречаются и у здоровых людей.
Если морфологические данные недостаточны для окончательного подтверждения МДС, показано проведение цитогенетических исследований. Хромосомные аномалии встречаются примерно в 30 – 50% случаев при первичном МДС и в 80 % – при вторичном МДС. Некоторые хромосомные аномалии ассоциируются с определенными клиническими проявлениями.
Например, потеря части длинного плеча 5 хромосомы связана с развитием макроцитарной анемии у пожилых женщин и с низким риском трансформации в острый миелолейкоз.
FAB-классификация
Классификация МДС представлена в табл. 2 .
Течение и прогноз
Клиническое течение МДС отличается большим разнообразием. Примерно две трети больных погибают вследствие недостаточности костного мозга. Медиана продолжительности жизни в целом составляет 20 мес. Прогноз для каждого пациента определяется количеством бластов в костном мозге, количеством и особенностями хромосомных аномалий и количеством заинтересованных ростков кроветворения.
Большинству пациентов может быть предложено лишь паллиативное лечение. Однако в отношении больных молодого возраста, перспективных в плане аллогенной трансплантации костного мозга, возможно полное излечение. Пятилетняя выживаемость в этих случаях составляет 40% при изначально повышенном количестве бластов в костном мозге и 60% – если такого повышения нет.
При низком риске показано наблюдение. При среднем или высоком риске для лиц моложе 65 – 70 лет перспективной является интенсивная химиотерапия. Ремиссия достигается примерно в 60 % случаев, но ее продолжительность редко превышает 18 мес.
Возможно, что широкое распространение для лечения МДС получат препараты, способствующие дифференцированию незрелых клеток (цитозар в низких дозах, аналоги витамина D3, ретиноевая кислота).
Для повышения содержания кровяных клеток применяют переливание препаратов крови. В этих же целях могут быть использованы различные колониестимулирующие факторы. Однако эти препараты дорогостоящи и неэффективны при тяжелых цитопениях.
Oscier DG The myelodysplastic syndromes BMJ 1997;314:883–6.
Миелодиспластические синдромы (МДС) — биологически и клинически гетерогенная группа клональных заболеваний, характеризующихся дисплазией кроветворения с неэффективным гемопоэзом и цитопеническими синдромами периферической крови и различной вероятностью эволюции в острые миелоидные лейкозы.
Признаки дисплазии кроветворения нередко сопровождаются бластозом крови и костного мозга, однако количество бластных клеток всегда меньше 20% (при уровне бластов равном или более 20% ставится диагноз острого лейкоза). В медицинской литературе прежних лет миелодиспластический синдром (МДС) имел различные названия (малопроцентный острый лейкоз, предлейкоз, тлеющая лейкемия и др.).
Частота миелодиспластических синдромов (МДС) в популяции составляет 3-5 случаев на 100 000 населения в год и существенно увеличивается с возрастом (частота у лиц старше 70 лет достигает 20 случаев на 100 000 населения в год). Средний возраст больных на момент начала заболевания составляет 70 лет.
Этиология и патогенез миелодиспластических синдромов
К этиологическим факторам миелодиспластического синдрома (МДС) относятся ионизирующее излучение, цитостатические препараты, производные бензола и другие химические агенты (в том числе продукты табакокурения), генетические факторы.
Предшествующее лечение онкологических и онкогематологических заболеваний с использованием радиологических методов, алкилирующих средств (хлорамбуцил, циклофосфамид, мельфалан) и эпиподофиллотоксинов (этопозид, тенипозид) существенно повышает риск развития МДС, причем пик заболеваемости после лечения алкилирующими агентами отмечается через 5-10 лет, после эпиподофиллотоксинов — через 5 лет.
Длительное использование алкилирующих препаратов при лечении ревматических или других неопухолевых заболеваний также сопровождается высоким риском развития миелодиспластического синдрома (МДС). У детей с синдромом Швахмана-Дайемонда, анемией Фанкони и нейрофиброматозом 1-го типа частота развития миелодиспластического синдрома (МДС) выше, чем в общей популяции.
Как и другие гемобластозы, миелодиспластический синдром (МДС) имеет клональный патогенез. Родоначальницей клона является дефектная стволовая кроветворная клетка. В развитии заболевания имеют значение дефекты кроветворного микроокружения, приводящие к нарушению продукции цитокинов клетками стромы костного мозга и сопровождающиеся кумуляцией хромосомных повреждений и нарушением регуляции апоптоза.

Клиническая картина миелодиспластических синдромов
Симптоматика миелодиспластического синдрома (МДС) обусловлена наличием и выраженностью цитопении (чаще встречается панцитопения, реже — одно- и двуростковая цитопения). Основные клинические синдромы: анемический, геморрагический и инфекционных осложнений.
Наиболее часто первым признаком заболевания является анемия, проявляющаяся общей слабостью, одышкой при обычной физической нагрузке, головокружениями, сердцебиениями. Геморрагический синдром обусловлен тромбоцитопенией и качественными нарушениями клеток мегакариоцитарного ростка и обычно манифестирует подкожными кровоизлияниями, носовыми и десневыми кровотечениями, меноррагиями у женщин.
При глубокой тромбоцитопении могут возникать менометроррагии, желудочно-кишечные, почечные кровотечения, острые нарушения мозгового кровообращения. У 20-30% больных в клинической картине преобладают инфекционные осложнения, частота и выраженность которых связаны со степенью и длительностью нейтропении или агранулоцитоза: в более легких случаях возникают стрептодермии, стоматиты, синуситы, в тяжелых случаях — пневмонии, сепсис, причем возбудителями инфекционных осложнений часто являются условно-патогенная бактериальная, вирусная и грибковая микрофлора.
Увеличение лимфатических узлов, печени и селезенки наблюдается у 10-20% больных. Исключением является хронический миеломоноцитарный лейкоз, при котором спленомегалия отмечается почти у половины пациентов.
У 10% больных в начале заболевания клинические признаки отсутствуют и миелодиспластический синдром (МДС) обнаруживается случайно (при исследовании крови).
В 10-50% случаев (в зависимости от варианта миелодиспластического синдрома (МДС)) в исходе заболевания развиваются вторичные острые миелоидные лейкозы. В связи с резистентностью к цитостатической терапии и пожилым возрастом большинства пациентов ремиссии достигаются редко и обычно непродолжительны.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Миелодиспластический синдром (МДС) – это не одна какая-то болезнь, это целая группа различных патологических состояний костного мозга (КМ), отнесенных к гематологии, но пока не причисленных к лейкозам, хотя болезнь оставляет высокий риск перехода в более тяжелую форму (лейкоз).
Суть МДС заключается в нарушении костномозгового кроветворения на миелоидной линии в отношении какого-то одного клона клеток или затрагивающего несколько популяций. В любом случае для миелодиспластического синдрома характерным признаком будет изменение качественного и количественного состава периферической крови.
Коротко о гемопоэзе
- Красных кровяных телец – эритроцитов;
- Белых клеток – лейкоцитов;
- Кровяных пластинок (бляшек Биццоцеро) – тромбоцитов.
Кроветворение начинается от стволовой клетки, способной, дифференцироваться и давать жизнь всем линиям (росткам) гемопоэза. Миелоидный и лимфоидный ростки пошли от специализированных, обладающих высокой пролиферативной активностью, способных к дифференцировке плюрипотентных клеток.

Сбой кроветворения в миелоидном направлении приводит к тому, что сам аномальный клон в некоторой степени теряет возможность продолжать линию (воспроизводить потомство, поэтому количество клеток того ростка, на котором возникла проблема, падает). Естественно, нарушается и созревание полноценных клеток. В результате этого, уменьшается численность одной или нескольких популяций форменных элементов, а также, ввиду ухудшения качества клеток, не в лучшую сторону меняются их функциональные возможности.
Вытекающие из подобных событий последствия – синдром, имеющий различные варианты клинических проявлений, то есть, представляющий собой группу гетерогенных патологических состояний, которая и названа миелодиспластическим синдромом.
Позиция МДС в Международной классификации болезней
Международная классификация болезней десятого пересмотра (МКБ-10), принятая Всемирной организацией здравоохранения (ВОЗ) в Швейцарии Женева, 1989), вступила в силу на территории Российской Федерации в 1997 году. Между тем, в отношении многих патологических состояний в 2010 году были внесены изменения. Нововведения коснулись и гематологической патологии, в том числе, миелодиспластического синдрома. По МКБ-10 в блок диагнозов D37-D48 МДС входит под своим кодом – D46, который имеет 7 или 9 вариантов определений заболеваний или диагнозов (в России, наряду с классификацией ВОЗ, могут использоваться и другие классификации, например, FAB, где вообще только 5 вариантов, поэтому в разных справочниках кодирование также может иметь отличия):
-
D0 Рефрактерная анемия (РА) без сидеробластов, так обозначенная (в периферической крови – анемия, бластов нет, в КМ – дисплазия, затрагивающая преимущественно эритроцитарный росток, 20% по данным ВОЗ или >30% согласно классификации FAB), то гематологи склоняются к диагнозу – миелоидный лейкоз. В ситуациях, когда численность бластных клеток вплотную не подходит к этому порогу, диагноз пациента остается прежним – миелодиспластический синдром.
Патологическое состояние главного кроветворного органа может сформироваться у человека в любом возрастном периоде (от грудного – до глубокой старости). У детей болезнь чаще всего дебютирует между 3 и 5 годами, хотя, в целом, в детском возрасте риск заболеть совсем низкий. Среди взрослых самыми уязвимыми становятся пожилые люди (60 лет и старше). Например, такой распространенной и рискующей перейти в острый лейкоз форме, как РЦМД, наиболее подвержены люди в возрасте от 70 до 80 лет. Общая частота встречаемости миелодиспластического синдрома колеблется в пределах 3-5 случаев на 100 тысяч населения (не так и редко), причем, мужчины страдают данной патологией несколько чаще, нежели женщины.
- Воздействие ионизирующего излучения;
- Влияние антропогенных неблагоприятных факторов окружающей среды (химических соединений, созданных человеком);
- Последствия химио- и радиотерапии (после лечения опухолевых процессов);
- Инфекционные агенты (бактерии, вирусы).
Следует отметить, что до сих пор МДС, передаваемого по наследству или возникающего в кругу близких родственников, отмечено не было, однако из наблюдений определена группа пациентов, имеющих повышенный риск формирования синдрома. Это дети и взрослые люди, страдающие болезнью Дауна, анемией Фанкони, синдромами Луи-Бар и Блума.
Лечатся все по-разному

Следует сразу настроить пациента, что лечение МДС не будет одинаковым для всех его разновидностей. Набор терапевтических мероприятий рассматривается в индивидуальном порядке, исходя из формы болезни и категории риска, которой принадлежит пациент (согласно клинической классификации Международной Прогностической Системы – IPSS для МДС: низкий, промежуточный 1 и 2, высокий). Словом, существуют определенные каноны, которых доктор придерживается, прежде чем приступить к непосредственному лечению. К примеру:
- Люди, не перешагнувшие 60-летний рубеж, имеющие минимальные признаки болезни, но отнесенные к категории промежуточного или высокого риска с ожидаемой выживаемостью 0,3 – 1,8 года, подвергаются высокоинтенсивной терапии;
- Пациенты, принадлежащие к группе промежуточного и низкого риска с ожидаемой выживаемостью 5-12 лет, проходят лечение низкой интенсивности;
- Молодые люди и больные среднего возраста (до 60 лет) с относительно неплохими показателями (ожидаемая выживаемость от полугода до 5 лет) первоначально лечатся по схемам низкой интенсивности, хотя в любой момент им грозит оказаться в группе, получающей более жесткое лечение (высокие дозы химиотерапии, пересадка КМ).
Таким образом, схемы лечения миелодиспластического синдрома довольно сложны и знает их только врач, получивший в свое время определенную специализацию (гематолог). Он в своей лечебной тактике опирается на рекомендации, разработанные Британским комитетом по стандартизации в гематологии (редакция 2009 года). Читателю же, на наш взгляд, достаточно познакомиться с основными методами проведения терапевтических мероприятий, особо не вникая в тонкости, не ставя диагноз и не причисляя себя или своих близких к той или иной группе риска. И еще, наверное, не помешает знать, что:
- Лечение высокой интенсивности – это, во-первых, обязательное пребывание в специализированном стационаре, во-вторых, назначение высоких доз химиотерапии и, возможно, подготовка к пересадке стволовых клеток и сама пересадка;
- Низкоинтенсивная терапия подразумевает пребывание в больнице (или даже в условиях дневного стационара) время от времени для получения заместительной терапии, низких доз химиопрепаратов, симптоматического лечения.
К сожалению, способа избавиться от такого тяжелого недуга, как МДС, раз и навсегда, пока не придумали. Разве что пересадка главного кроветворного органа (костного мозга) могла бы решить проблему, однако она тоже сопряжена с определенными трудностями (иммунологическое типирование, поиск совместимого донора, высокая стоимость операции, если искать донора по всему миру). Правда, в последние годы, как на территории Российской Федерации и ближайшей соседки – Беларуси, так и на территории других государств бывшего СССР, создаются новые лаборатории тканевого типирования, объединяющие свои реестры в единый банк, чтобы иметь возможность помочь друг другу. На них и возлагаются будущие надежды.
Лечение

Если врач считает, что патологический процесс идет как бы доброкачественно (если можно так выразиться), с небольшим количеством бластов, то больные группы низкого риска, периодически получающие заместительное и поддерживающее лечение (эритроцитарную массу, тромбовзвесь), могут довольно продолжительное время работать и вести почти привычный образ жизни. В основном, лечение таких больных выглядит следующим образом:
Пересадка стволовых клеток (возможна тоже только до 60 лет) на сегодняшний день – единственный способ избавить человека от страданий на долгие-долгие годы. К сожалению, трансплантация КМ – операция хоть и несложная в техническом плане, но трудновыполнимая в плане подбора по лейкоцитарной системе HLA совместимого с реципиентом (больным) донора (идентичными, то есть, имеющими абсолютно одинаковый набор генов являются только однояйцевые близнецы – это идеальные доноры друг другу).
Частные симптомы и диагностика
Клинические проявления и степень их выраженности по причине многообразия форм МДС позволяют себе широкие вариации. Случайной находкой синдром выступает редко (это бывает, если человек неплохо себя чувствует, а анализы назначаются в силу других обстоятельств). В основном же, больные направляются в поликлинику с определенными жалобами (постоянное ощущение усталости, одышка, физическая слабость, головокружения, частые подъемы температуры тела), где после тестирования крови становятся очевидными и другие признаки миелодиспластического синдрома:
- Цитопения (снижение количества полноценных форменных элементов крови);
- Анемия (низкий гемоглобин, мало эритроцитов), которая и определяет симптомы, заставившие пойти к врачу;
- Нейтропения (недостаточное содержание в крови нейтрофильных лейкоцитов, обладающих способностью поглощать бактериальные клетки в очаге воспаления – она становится причиной частых инфекций и лихорадки);
- Тромбоцитопения (уменьшение численности тромбоцитов, что обуславливает появление геморрагического синдрома – кровотечений, мелкоточечных подкожных кровоизлияний, синяков).
Чаще всего поводом все же обратиться в поликлинику служат жалобы больного, которые в наибольшей степени связаны с анемией. Пробовать повысить уровень красного пигмента крови (Hb) и содержание красных кровяных телец (Er) препаратами железа и витаминами бесполезно, лечение успехов не приносит, ведь анемия при МДС – рефрактерная. При подозрении на МДС, которое возникает в ходе проведения общего анализа крови (ОАК), добавляются другие исследования:
трепанбиопсия костного мозга
Трепанобиопсия (процедура обязательна для всех больных) – после изучения морфологических особенностей гистологический анализ развеет сомнения или подтвердит подозрения;
Безусловно, диагностика миелодиспластического синдрома, начинается с жалоб больного и ОАК, но в дальнейшем опирается на более сложные лабораторные исследования. Здесь врачу есть над чем подумать, чтобы правильно оценить нарушения кроветворения, ведь изменения клеточного состава и морфологических особенностей клеток крови и костного мозга могут быть весьма многочисленны и многообразны. Впрочем, как и сама болезнь…

костный мозг при МДС
Прогноз и рекомендации
Прогноз в отношении продолжительности жизни при миелодиспластическом синдроме не очень оптимистичный, хотя многое зависит от разновидности болезни, степени риска и возрастной категории больного. В целом, пациенты, строго выполняющие рекомендации лечащего врача и получающие периодически поддерживающее лечение, могут рассчитывать прожить пять, а то и десять лет. Однако активное течение злокачественной формы болезни оставляет мало шансов – если не был найден донор и не пересажена стволовая клетка, жизнь может прерваться на 1-2 году от начала патологического процесса. Причиной смерти в большинстве случаев становится острый миелоидный лейкоз, который развился на почве вторичного МДС.
Миелодиспластический синдром (МДС) сегодня является одной из самых сложных проблем гематологии. Лишь недавно лечение МДС вышло за рамки поддерживающей терапии, проводившейся с помощью облегчения симптомов. МДС является патологией старшей возрастной группы: 80 % случаев МДС приходится на лица старше 60 лет. МДС в детском возрасте встречается крайне редко. В европейских странах среди лиц 50—69 лет регистрируется 40 новых случаев МДС на 1 млн населения, а среди лиц 70 лет и старше — 150 новых случаев на 1 млн населения. Заболеваемость МДС в РФ в среднем составляет 3—4 случая на 100 тыс. населения в год и увеличивается с возрастом [1].

1.Рукавицын О. Гематология: национальное руководство / О. Рукавицын. – Москва: ГЭОТАР-Медиа, 2017. –С. 193-226.
2. Герминг У. Миелодиспластические синдромы: диагностика, прогноз, лечение / У. Герминг, Г. Коббе, Р. Хаас // Deutsches Ärzteblatt International. -2013. -№110(46). -90 с.
4. Иванага М. Риск миелодиспластических синдромов у людей, подвергшихся воздействию ионизирующего излучения: ретроспективное когортное исследование людей, переживших атомную бомбу Нагасаки / М. Иванга, М. Сода, Ю. Такасаки [и др.] // Журнал клинической онкологии. -2011. -№29(4). С. 34-42.
5. Каззола М. Экспрессия митохондриального ферритина в эритроидных клетках пациентов с сидеробластной анемией / М. Каззола, Р. Инверниззи, Дж. Бергамаши, С. Леви [и др.] // Журнал Кровь. -2003. -№101(5). –С. 1996-2000.
7. Бхатнагар Н. Транзиторный аномальный миелопоэз и острый миелоидный лейкоз при синдроме Дауна / Н. Бхатнагар, Л. Низери, О. Танстолл, П. Вьяс, И. Робертс // Текущие гематологические отчеты о злокачественных заболеваниях. -2016. -№11(5). –С. 33-41.
МДС – группа заболеваний со сложным патогенезом, который приводит к развитию диспластического кроветворения в сочетании с нормальным. Вначале симптомы обычно не проявляются. Позже симптомы могут включать чувство усталости, одышку, нарушения свертываемости крови, анемию или частые инфекции. Некоторые типы могут перерасти в острый миелоидный лейкоз [2].
Вторичный МДС является значительно более неблагоприятным и резистентным к лечению типом МДС, обладающим заведомо более худшим прогнозом в сравнении с первичным МДС.
Признаки и симптомы неспецифичны и обычно связаны с цитопенией крови: анемия (хроническая усталость, одышка, ощущение холода, иногда боль в груди); нейтропения (повышенная восприимчивость к инфекции); тромбоцитопения (повышенная склонность к кровотечениям и экхимозам, а также к подкожным кровотечениям, приводящим к пурпуре или петехиям) [3].
Многие люди не имеют симптомов, и цитопения крови или другие проблемы выявляются при обычном анализе крови: нейтропения, анемия и тромбоцитопения; спленомегалия или редко гепатомегалия; аномальные гранулы в клетках, аномальная форма и размер ядер; хромосомная аномалия, включая хромосомные транслокации и аномальное количество хромосом
Хотя существует определенный риск развития острого миелоидного лейкоза, около 50% смертей происходит в результате кровотечения или инфекции. Однако лейкоз, возникающий в результате миелодисплазии, обычно не поддается лечению. На раннем этапе преобладает анемия. Большинство пациентов жалуются на постепенное наступление утомляемости и слабости, одышки и бледности, но, по крайней мере, у половины пациентов симптомы отсутствуют, и МДС обнаруживается лишь случайно при обычных анализах крови. Предшествующая химиотерапия или облучение являются важным фактором в истории болезни человека. Лихорадка и потеря веса должны указывать на миелопролиферативный, а не на миелодиспластический процесс. [3]
МДС чаще всего развивается без видимой причины. Факторы риска включают воздействие агента, который, как известно, вызывает повреждение ДНК, такого как радиация, бензол и некоторые виды химиотерапии; о других факторах риска сообщалось непоследовательно. Доказать связь между предполагаемым воздействием и развитием МДС может быть сложно, но наличие генетических аномалий может предоставить некоторую подтверждающую информацию. Вторичный МДС может возникать как поздняя токсичность раковой терапии (МДС, ассоциированный с терапией, t-МДС). МДС после воздействия радиации или алкилирующих агентов, таких как бусульфан, нитрозомочевина или прокарбазин, обычно возникает через 3-7 лет после воздействия и часто демонстрирует потерю хромосомы 5 или 7. МДС после воздействия ингибиторов ДНК-топоизомеразы II возникает после более короткого латентного периода - всего 1–3 года и может иметь транслокацию 11q23. Другие ранее существовавшие заболевания костного мозга, такие как приобретенная апластическая анемия после иммуносупрессивного лечения и анемия Фанкони, могут перерасти в МДС.
Считается, что МДС возникает из-за мутаций в мультипотентных стволовых клетках костного мозга, но конкретные дефекты, ответственные за эти заболевания, остаются плохо изученными. Дифференциация клеток-предшественников крови нарушается, и в клетках костного мозга происходит значительное увеличение уровней апоптотической гибели клеток. Клональная экспансия аномальных клеток приводит к образованию клеток, утративших способность дифференцироваться. Если общий процент миелобластов костного мозга превышает определенный предел (20% для ВОЗ), то считается, что произошла трансформация в острый миелогенный лейкоз (ОМЛ). Прогрессирование МДС в ОМЛ - хороший пример многоэтапной теории канцерогенеза, в которой серия мутаций происходит в изначально нормальной клетке и превращает ее в раковую клетку.
Хотя признание лейкемической трансформации было исторически важным, значительная часть заболеваемости и смертности, связанных с МДС, является результатом не трансформации в ОМЛ, а, скорее, цитопений, наблюдаемых у всех пациентов с МДС. В то время как анемия является наиболее распространенной цитопенией у пациентов с МДС, учитывая доступность переливания крови, пациенты с МДС редко страдают от тяжелой анемии. Двумя наиболее серьезными осложнениями у пациентов с МДС в результате их цитопении являются кровотечение (из-за недостатка тромбоцитов) или инфекция (из-за недостатка лейкоцитов). Длительное переливание эритроцитов приводит к перегрузке железом.
Признание эпигенетических изменений в структуре ДНК при МДС объяснило успех двух (а именно гипометилирующих агентов 5-азацитидин и децитабин) из трех (третий - леналидомид) коммерчески доступных лекарств, одобренных Управлением по контролю за продуктами и лекарствами США для лечения МДС. Правильное метилирование ДНК имеет решающее значение для регуляции генов пролиферации, а потеря контроля метилирования ДНК может привести к неконтролируемому росту клеток и цитопении. Недавно одобренные ингибиторы ДНК-метилтрансферазы используют этот механизм, создавая более упорядоченный профиль метилирования ДНК в ядре гемопоэтических стволовых клеток, тем самым восстанавливая нормальные показатели крови и замедляя прогрессирование МДС до острого лейкоза [3].
Некоторые авторы предположили, что потеря митохондриальной функции с течением времени приводит к накоплению мутаций ДНК в гемопоэтических стволовых клетках, и это объясняет повышенную частоту МДС у пожилых пациентов. Исследователи указывают на накопление митохондриальных отложений железа в кольцевых сидеробластах как на доказательство митохондриальной дисфункции при МДС [5].
По крайней мере, с 1974 г. известно, что делеция в длинном плече хромосомы 5 связана с диспластическими аномалиями гемопоэтических стволовых клеток. К 2005 году леналидомид, химиотерапевтический препарат, был признан эффективным у пациентов с МДС с 5q-синдромом, а в декабре 2005 года FDA США одобрило этот препарат для этого показания. Пациенты с изолированным 5q-, низким риском IPSS и трансфузионной зависимостью лучше всего реагируют на леналидомид. Как правило, прогноз для этих пациентов благоприятный, средняя выживаемость составляет 63 месяца. Леналидомид имеет двойное действие, снижая количество злокачественных клонов у пациентов с 5q- и индуцируя лучшую дифференцировку здоровых эритроидных клеток, как это наблюдается у пациентов без делеции 5q [6].
Мутации в факторах сплайсинга были обнаружены в 40-80% случаев миелодиспластического синдрома, особенно у пациентов с кольцевидными сидеробластами.
Мутации в генах, кодирующих изоцитратдегидрогеназу 1 и 2 (IDH1 и IDH2), встречаются у 10–20% пациентов с миелодиспластическим синдромом и приводят к ухудшению прогноза при МДС низкого риска. Поскольку частота мутаций IDH1 / 2 увеличивается по мере увеличения злокачественности заболевания, эти данные вместе предполагают, что мутации IDH1 / 2 являются важными факторами прогрессирования МДС в более злокачественное состояние.
Преходящее миелопролиферативное заболевание - аномальная пролиферация клона доброкачественных мегакариобластов в печени и костном мозге. Заболевание ограничивается людьми с синдромом Дауна или генетическими изменениями, аналогичными таковым при синдроме Дауна, развивается во время беременности или вскоре после рождения и проходит в течение 3 месяцев, или примерно в 10% случаев прогрессирует до острого мегакариобластного лейкоза [7].
Читайте также: