
Наследственные нарушения обмена углеводов реферат
Обновлено: 04.07.2024
Наследственные нарушения углеводного обмена, ведущие к поражению печени
Авторы: К.Н. Бородий, Донецкий национальный медицинский университет им. М. Горького
Версия для печати
К патологии печени приводит гетерогенная группа наследственных болезней, обусловленных различными видами нарушения обмена углеводов. Различают:
— нарушения обмена моно- и ди-сахаридов;
— болезни накопления — гликогенозы;
— патология соединительной ткани — мукополисахаридозы;
— другие.
Нарушения обмена моно- и дисахаридов
Фруктоземия
Этиология и патогенез. Заболевание обусловлено врожденным отсутствием ферментов фруктозофосфатальдолазы и фруктозодифосфатальдолазы. Избыточное накопление фруктозофосфата нарушает гликогенолиз, что приводит к гипогликемии. В печени имеется недостаточное количество фермента фруктозо-1-фосфат-альдолазы, в результате продукты обмена (фруктозо-1-фосфат) накапливаются в организме (печени, почках, слизистых оболочках кишечника) и оказывают повреждающее действие. Морфологически в печени выявляются жировая инфильтрация, умеренный перилобулярный фиброз.
Клиническая картина. Симптомы возникают при введении в рацион сладкой пищи или фруктовых соков, т.е. продуктов, содержащих фруктозу. Со 2–4-го месяца развиваются диспептические явления и состояния острой гипогликемии, которые проявляются бледностью, вялостью, потливостью, запахом ацетона. В тяжелых случаях может развиться гипогликемическая кома с потерей сознания и судорогами. Характерно, что гипогликемия возникает после приема пищи. С возрастом дети сами отказываются от сладкой пищи. Постоянным признаком является гепатомегалия (обычно с увеличением обеих долей) с ровным краем и некоторым уплотнением печени.
Лечение. Диета, лишенная фруктозы, главными источниками которой считаются мед, сахарный тростник и свекла, фрукты, джемы, повидло, морковь, какао, цикорий, репа. Больным разрешается употреблять молоко и молочные продукты, яйца, маслины, подсолнечное масло, животные жиры. Разрешается женское и коровье молоко. Сухое молоко должно быть без сахара (сахарозы). Допускаются все виды сыров и натуральные кисломолочные продукты (неподслащенные). Запрещается молоко с добавлением сахарозы, сгущенное молоко, подслащенные кисломолочные продукты. Разрешены мясо и рыба. Исключаются колбасы и колбасные изделия, консервы. Жиры включаются в диету практически без ограничений (сливочное и растительное масло, маргарин). Почти все фрукты запрещены. Можно употреблять в пищу лимоны и каштаны. Из овощей допускаются зеленые бобы, кресс-салат, латук, лук-порей, капуста, шпинат. Разрешаются натуральная пшеничная или ржаная мука, рис, хлеб, манная крупа, чай, кофе, какао без сахара, глюкоза, мальтоза, декстрин-мальтоза, сахарин. Запрещаются соя, мука с сахарозой, бисквиты, пирожные, лимонад и все газированные фруктовые напитки, соки, сиропы, сахар, варенье, нуга, конфеты, все медикаменты, содержащие сахар, сорбитол (гранулы, драже, порошки, пилюли).
Новорожденным назначают молочные смеси без сахара. Дети первого года получают молочные смеси, содержащие только лактозу и декстрин-мальтозу. Вместо фруктовых пюре и соков питание дополняют глюкозой (от 30 до 60 г ). При введении прикорма раньше, чем здоровым детям, назначают мясо, рыбу, сыр, яйца. Аскорбиновую кислоту используют без сахара. Диетическое питание показано до 5–6 лет, только по достижении этого возраста можно в ограниченных количествах, под контролем врача и биохимических анализов крови, включать в питание ребенка продукты из запрещенного списка.
Галактоземия
Частота 1 : 35 000–50 000 населения.
Этиология и патогенез. Наследственная энзимопатия. Наследуется по рецессивному типу. В основе галактоземии (рис. 1) лежит нарушение обмена галактозы в связи с отсутствием фермента галактозофосфат-уридилтрансферазы. В результате в крови накапливаются в больших концентрациях галактоза и галактозофосфат. Происходит нарушение процесса ферментативного превращения галактозы в глюкозу с накоплением галактозы и продуктов ее обмена в клетках, что оказывает повреждающее действие на функции печени, головного мозга, хрусталика глаза, почек.
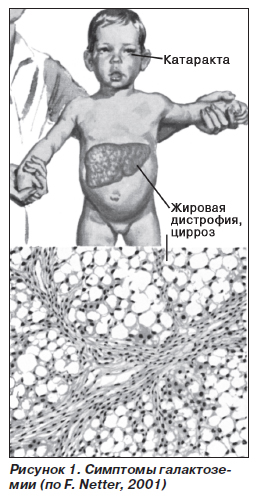
Клиническая картина. Клинические признаки заболевания возникают рано — через 1–2 недели после рождения ребенка. Пропадает аппетит, появляются вялость, рвота, понос. Наблюдается дефицит массы тела. Постепенно развивается гепато-, спленомегалия, появляется стойкая гипербилирубинемия, преимущест-венно за счет прямого билирубина. Часто отмечается катаракта, ведущая к слепоте. Могут быть симптомы, свидетельствующие о поражении почек (протеинурия, гипераминоацидурия), центральной нервной системы (задержка психофизического развития). После чайно-водной паузы состояние улучшается, но введение молока обусловливает рецидив нарушений со стороны желудочно-кишечного тракта. При несвоевременной диагностике заболевание прогрессирует, что приводит к тяжелым последствиям или летальному исходу.
Диагностика:
1. Определение концентрации га-лактозо-1-фосфата в эритроцитах (повышена).
2. Исследование активности галак-тозо-1-фосфат-уридилтрансферазы в эритроцитах.
3. Повышение уровня галактозы в крови и моче (методом хроматографии).
4. Микробиологический тест Гатри.
Лечение. Диетотерапия является единственным методом лечения. Для вскармливания ребенка используют смеси, лишенные лактозы. Из питания детей более старшего возраста исключают цельномолочные продукты.
Гликогенозы
Этиология и патогенез. Группа наследственных болезней обмена полисахаридов, развивающихся в результате нарушения синтеза или распада гликогена на простые сахара. При этом нормальный и аномальный гликоген одновременно накапливаются в клетках печени и других органах и тканях.
Гликоген — это сильно разветвленный полимер глюкозы, в котором большинство остатков имеют 1,4-связи, а 7–10 % остатков — 1,6-связи. Древовидная структура подвергается надстройке и отщеплению остатков на периферии молекулы. Молекулярная масса гликогена составляет несколько миллионов, его молекулы могут агрегировать с образованием структур, видимых при электронной микроскопии. В печени гликогена обычно содержится менее 70 мг/г, а в мышцах — менее 15 мг/г, но эти величины колеблются в зависимости от питания и гормональных влияний. Нарушения структуры гликогена могут быть связаны как с уменьшением, так и с увеличением ветвления молекулы.
Глюкоза мобилизуется из гликогена целым рядом ферментативных реакций. На гликоген непосредственно действует активная форма фосфорилазы — фосфорилаза а, отщепляя отдельные остатки глюкозы и образуя Г-1-Ф. В печени и мышцах фосфорилаза кодируется разными генными продуктами. В этих тканях фермент может существовать в активной фосфорилированной и неактивной дефосфорилированной формах. Фосфорилаза представляет собой димер, состоящий из одинаковых субъединиц, причем обе формы фермента подвергаются сложной аллостерической регуляции. Неактивная фосфорилаза b превращается в активную форму под действием фосфорилазо-b-киназы, которая существует и в активной фосфорилированной и неактивной дефосфорилированной формах. Этот фермент состоит из четырех разных субъединиц (a, b, g, d4),причем d-цепь идентична связывающему кальций белку — кальмодулину. Скорость мобилизации глюкозы этой системой регулируется каскадом киназных реакций, включающих цАМФ.
По характеру ферментной недостаточности принято различать 12 типов гликогенозов, среди которых выделяют печеночные (гликогенозы 0, I, III, IV, VI, VIII, IX, Х, ХI типов) или мышечные формы (гликогенозы V и VII типов). Гликогеноз II типа проявляется поражением только мышц или поражением многих систем и органов (генерализованная форма). Также возможны сочетания нескольких типов.
Клиническая картина гликогенозов характеризуется гипогликемией (рвота, судороги, потеря сознания, кома). Течение болезни зависит от места депонирования гликогена: печень, почки, мышечная ткань. Соответственно выделяют цирроз печени, почечную форму, мышечный синдром (включая сердечную форму). Преобладание у новорожденного ребенка симптомов гипогликемии может привести к синдрому внезапной смерти. Прогноз зависит от типа болезни.
Классификация основана на различиях в дефектах ферментов, лежащих в основе заболеваний.
Гликогеноз 0 типа (агликеноз) характеризуется резким снижением запасов гликогена в печени, наблюдается тяжелое состояние вплоть до развития комы (гипогликемический синдром). Кома может возникать после рождения при позднем прикладывании ребенка к материнской груди, а позднее — утром натощак и в перерывах между кормлениями. При отсутствии лечения ребенка наступает нарушение психомоторного развития.
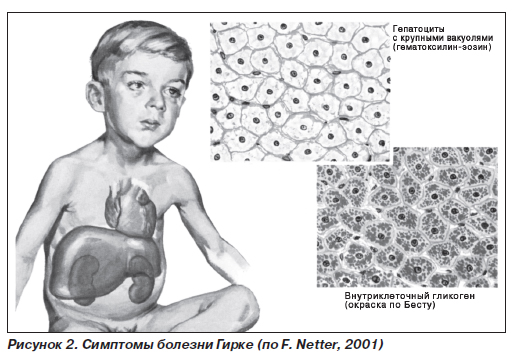
Гликогеноз I типа (болезнь Гирке) (рис. 2) — гликогеноз, обусловленный недостаточностью глюкозо- 6-фосфатазы, приводящей к невозможности превращения глюкозо-6-фосфата в глюкозу, что сопровождается накоплением гликогена в печени и почках; наследуется по аутосомно-рецессивному типу. Дефект фермента в печени, почках, слизистой оболочке тонкой кишки. При его первых проявлениях наблюдаются отсутствие аппетита, рвота, респираторный дистресс-синдром, гипогликемические судороги (кома), которые выявляются сразу после рождения или в грудном возрасте. Прогрессируют гепатомегалия и нефромегалия за счет гликогенной инфильтрации. С течением времени появляются: отставание в росте, диспропорция тела (большая голова, короткие шея и ноги), кукольное лицо, гипотония мышц; половое созревание задерживается. Нервно-психическое развитие удовлетворительное. В связи с резкой гипогликемией больные вынуждены постоянно принимать пищу.
Гликогеноз II типа (болезнь Помпе) — наследуется по аутосомно-рецессивному типу. Симптомы проявляются в первые недели жизни — до шести месяцев после рождения. Дефект фермента найден в печени, почках, селезенке, мышцах, нервной ткани, лейкоцитах. Наблюдается расстройство дыхания, беспокойство или адинамия. Отмечаются отсутствие аппетита, задержка роста, мышечная гипотония. Увеличиваются размеры сердца, печени, почек, селезенки. Сердце приобретает шаровидную форму, в связи с гипертрофией миокарда появляются изменения ЭКГ. Часто возникают гипостатические пневмонии, бронхиты, ателектазы легких, наблюдаются миодистрофия, гипорефлексия, спастические параличи. Мышечная форма гликогеноза II типа возникает только в мышцах при дефиците кислой α-1,4-глюкозидазы. Болезнь проявляется в более поздние сроки и по клинической картине напоминает миопатию.
Гликогеноз III типа (болезнь Кори, болезнь Форбса, лимитдекстриноз) — гликогеноз, вызванный недостаточностью фермента амило-1,6-глюкозидазы. Этот фермент катализирует расщепление связей […C-O-C…] в молекуле гликогена в точках ветвления. Болезнь сопровождается отложением атипичного гликогена в печени, сердце, мышцах. Наследуется по аутосомно-рецессивному типу. Дефект фермента найден в печени, мышцах, лейкоцитах, эритроцитах. С первых месяцев жизни ребенка наблюдаются гепатомегалия, мышечная гипотония, гипертрофия отдельных групп мышц. В некоторых случаях у больных отмечаются нарушение сердечной проводимости и кровообращения, гипертрофия миокарда. Развитие заболевания замедляется после пятилетнего возраста или в пубертатном периоде.
Гликогеноз IV типа (болезнь Андерсена, амилопектиноз, диффузный гликогеноз с циррозом печени) — гликогеноз, семейный цирроз печени, вызванный дефектом фермента амило-(1,4-1,6)-трансглюкозидазы. Этот фермент катализирует превращение 1,4-связей в молекуле гликогена в 1,6-связи, то есть обусловливает ветвление молекулы полисахарида. Заболевание сопровождается избыточным накоплением атипичного гликогена в печени. Наследуется по аутосомно-рецессивному или связанному с полом типу. Дефект фермента найден в печени, почках, мышцах, лейкоцитах. Болезнь наблюдается с первых месяцев жизни и характеризуется гепатоспленомегалией, развитием цирроза печени, желтухой, гипогликемией.
Гликогеноз V типа (болезнь Мак-Ардла, миофосфорилазная недостаточность) — гликогеноз, связанный с дефектом мышечной фосфорилазы. Заболевание, обусловленное нарушением каталитической функции этого фермента; сопровождается отложением значительного количества гликогена в мышцах. Наследуется по аутосомно-рецессивному типу. Дефект фермента найден в мышцах. В связи с гликогенной инфильтрацией скелетные мышцы увеличиваются в объеме, становятся очень плотными. Мышечная слабость, мышечные спазмы, тахикардия при физической нагрузке появляются в первые десять лет жизни и прогрессируют. Наблюдается транзиторная миоглобинурия. Концентрация лактата в крови после физической нагрузки уменьшается. Чаще (в 5 раз) болеют лица мужского пола.
Гликогеноз VI типа (болезнь Герса, гепатофосфорилазная недостаточность) — гликогеноз, вызванный недостаточностью фосфорилазы печени. Фосфорилаза печени катализирует фосфорилирование гликогена с образованием глюкозо-1-фосфата. Нарушение этого механизма приводит к избыточному отложению гликогена в печени. Наследуется, предположительно, по аутосомно-рецессивному типу. Проявляется обычно на первом году жизни. Характерны значительное увеличение печени в результате гликогенной инфильтрации гепатоцитов, задержка роста, кукольное лицо, гиперлипидемия, гипергликемия после внутривенного введения галактозы, повышенное содержание гликогена в эритроцитах. Наследуется по аутосомно-рецессивному типу. Дефект фермента найден в печени, лейкоцитах. Проявляется обычно на первом году жизни.
Гликогеноз VII типа (болезнь Таруи, миофосфофруктокиназная недостаточность) — симптомы сходны с гликогенозом V типа. Дефект фермента найден в мышцах, эритроцитах. Также характерны мышечная слабость, утомляемость и отсутствие гиперлактацидемии после физической нагрузки.
Гликогеноз IX типа (болезнь Хага) — наследуется по рецессивному, связанному с полом типу. Дефект фермента найден в печени. У больных наблюдается гепатомегалия.
Гликогеноз Х типа — известен случай у единственного больного, наследование не установлено. Дефект фермента найден в печени, мышцах. Наблюдалась гепатомегалия, через 6 лет после начала заболевания появились мышечные боли и спазмы мышц после физических упражнений.
Гликогеноз XI типа (болезнь Фанкони — Бикеля) — наследование не установлено. Дефект фермента найден в печени, почках. Характеризуется значительным увеличением печени и резкой задержкой роста. Наблюдаются симптомы гипофосфатемического рахита. В пубертатном периоде возможны уменьшение размеров печени, ускорение роста, нормализация уровня фосфора в крови.
Лечение гликогенозов в основном симптоматическое и направлено на изменение нарушений обменных процессов. Цель лечения — предупредить тяжелую гипогликемию. Назначают диету, богатую белками и углеводами. Питание должно быть частым (каждые 4 ч). Белки служат источником аминокислот — субстратов глюконеогенеза; они уменьшают углеводную нагрузку, приводящую к гипергликемии и лактат-ацидозу. Такая диета предотвращает гипогликемию и кетоацидоз натощак, уменьшает гипергликемию и лактат-ацидоз после еды и способствует ускорению роста. При мышечных формах гликогенозов улучшение отмечается при соблюдении диеты с высоким содержанием белка, назначении фруктозы, поливитаминов, АТФ. Иногда необходимо применение глюкагона, анаболитических гормонов, глюкокортикоидов. Предпринимаются попытки введения больным недостающих ферментов.
Профилактика не разработана.
Мукополисахаридозы
Этиология и патогенез. Мукополисахаридозы (МПС) — гетерогенная группа заболеваний, отнесенных к наследственным болезням обмена сложных сахаров. МПС сопровождаются избыточным накоплением в тканях и повышенной экскрецией гликоз-аминогликанов (ГАГ) — кислых мукополисахаридов, соединенных с белком и состоящих из уроновых кислот, аминосахароз и нейтральных сахаров. Указанные комплексы существуют в форме протеогликанов, являющихся важнейшими компонентами основного структурного белка волос (0-кератина) и структурного белка соединительной ткани (коллагена).
Для большинства МПС характерен аутосомно-рецессивный тип наследования, кроме синдрома Хантера (Х-сцепленный рецессивный).
Среди МПС выделяют ряд типов, каждый из которых обусловлен дефицитом специфической лизосомной гидролазы, участвующей в последовательном расщеплении ГАГ.
I тип — синдром Гурлера (4р16 — IDUA). Выделяют подтипы: Гурлер, Шейе, смешанный вариант. Для всех характерно снижение активности альфа-идуронидазы и накопление в тканях дерматан- и гепарансульфатов.
II тип — синдром Хантера (Гюнтера) (Хq28 — IDS). Снижение активности L-идуросульфат-сульфатазы и отложение в тканях дерматан- и гепарансульфатов. Клинические признаки менее выражены, продолжительность жизни больше, чем при других типах МПС.
III тип — синдром Санфилиппо (12q13.4). В зависимости от природы первичного биохимического дефекта выделяют четыре подтипа: А, В, С, D. В клинической картине преобладают психические расстройства: деменция, агрессивность. Продолжительность жизни не превышает 20 лет.
IV тип — синдром Моркио (16q24.3 — GALNS). Выделяют подтипы А и В. В тканях откладывается кератансульфат. Преобладают поражения скелета и непропорционально низкий рост.
V тип — синдром Шейе (см. I тип).
VI тип — синдром Марото — Лами (5q11.2 — ARSB). Дефицит фермента арилсульфатазы В. В тканях накапливается дерматансульфат. Фенотипически напоминает МПС I типа, но интеллект не снижен.
VII тип — синдром Слая (7q21.11 — GUSB). Дефицит фермента δ-глюкуронидазы. В тканях накапливаются дерматан-, гепаран- и хондроитин-сульфаты. Фенотипиче-ски напоминает МПС I типа, но имеет более доброкачественное течение.
Диагностика МПС основывается на совокупности данных генеалогического анализа, клинических проявлений, типичных рентгенологических данных, экскреции с мочой оксипролина (снижение), ГАГ и их фракций (превышение в 5–10 раз). Точная идентификация типов МПС возможна только с помощью определения активности лизосомных гидролаз в лимфоцитах и лейкоцитах крови, культуре фибробластов кожи, биоптатов печени, а также в моче.
Лечение больных МПС в основном симптоматическое, заключается в назначении терапии, способствующей нормализации или стабилизации патологического процесса в опорно-двигательном аппарате, сердечно-сосудистой и центральной нервной системах, паренхиматозных органах, органах зрения и слуха. Перспективным считается плазмаферез.
Участие углеводов в защитных реакциях иммунной системы организма. Рассмотрение наследстенных нарушений обмена углеводов. Причины и механизмы развития галактоземии. Признаки и лечение галактоземии у детей. Диагностирование и лечение фруктоземии.
| Рубрика | Медицина |
| Вид | реферат |
| Язык | русский |
| Дата добавления | 10.01.2017 |
| Размер файла | 25,2 K |
Студенты, аспиранты, молодые ученые, использующие базу знаний в своей учебе и работе, будут вам очень благодарны.
Кафедра биологической химии
Проверила: к.м.н. Газдалиева Л.М.
Выполнила: студентка МПФ 201Б
Файзулова Г.И.
Уфа 2014
Углеводы являются сложными органическими соединениями, выполняющими в организме человека ряд важнейших функций. Углеводы, состоящие из одной молекулы, называются моносахаридами; из двух связанных молекул -- дисахаридами; из нескольких молекул, связанных между собой, -- полисахаридами; состоящие из нескольких простых углеводов -- олигосахаридами. Основными моносахаридами организма человека являются глюкоза, фруктоза, галактоза и рибоза. Дисахариды и олигосахариды входят в состав разнообразных пищевых продуктов. Кроме того, в растительном и животном мире распространены полисахариды. Важнейшей функцией углеводов в человеческом организме является обеспечение энергией всех процессов, происходящих в нем. Углеводы используются и как строительный материал, являясь компонентом основного вещества соединительной, костной и хрящевой тканей. Они выполняют ряд специфических функций, в частности являются структурными элементами секрета желез человеческого организма (например, слюнных). Такие углеводы, как рибоза и дезоксирибоза, являются основным компонентом РНК и ДНК соответственно. Такое вещество, как глюкуроновая кислота, является производным глюкозы и участвует в процессе обезвреживания разнообразных токсических веществ в организме, а углевод гепарин участвует в процессе свертывания крови. Участие углеводов доказано в защитных реакциях иммунной системы организма. В организм человека с пищей поступают олиго- и полисахариды, которые в данном виде не могут усвоиться. В этой связи они подвергаются расщеплению на моносахариды под влиянием пищеварительных соков в желудочно-кишечном тракте. В таком виде они и подвергаются всасыванию в кишечнике, затем попадают в кровоток, оттуда переносятся в печень, где подвергаются дальнейшим химическим превращениям или накапливаются в ткани органа в качестве запасов. Углеводы принимают участие в обмене веществ, превращаясь, если это необходимо, в аминокислоты или жиры.
К наследственным нарушениям обмена углеводов относятся различные патологические состояния, обусловленные неспособностью организма усваивать углеводы или катаболизировать их. Эти формы патологии обмена часто проявляются в раннем возрасте. Патогенез поражения нервной системы при наследственных заболеваниях обмена углеводов связан с развитием частых гипер- и гипогликемических состояний, нарушением электролитного и водного баланса, с образованием токсических продуктов метаболизма (кетокислот и др.), вызывающих метаболические и структурные нарушения в ткани мозга, с дегенерацией клеток вследствие накопления в них некатаболизируемых углеводов.
Причины и механизмы развития галактоземии. Причина заболевания связана с тем, что организм ребенка не может усваивать сахар галактозу, который содержится в молоке. В итоге развиваются тяжелые нарушения со стороны печени, головного мозга, глаз и других органов. Болезнь встречается нечасто. Возможны случаи носительства патологического гена, в этом случае заболевание никак не проявляется, но в дальнейшем может возникать у потомков. Существуют три различные формы заболевания: обычная, форма, открытая Дюарте, и негритянская форма. В общем клинические проявления трех вариантов различаются только по степени выраженности. В результате повреждения генетического аппарата нарушаются превращения в организме сахара галактозы, накапливаются вредные продукты обмена веществ, что приводит к повреждению различных органов и тканей. Снижается содержание глюкозы в крови, вследствие чего такие дети очень медленно растут и плохо развиваются. Вредные продукты обмена накапливаются также и в нервной системе, приводя к отеку головного мозга и появлению неврологических отклонений. Часто патологические изменения приводят к развитию малокровия вследствие разрушения эритроцитов. В ряде случаев заболевание может протекать без нарушений со стороны печени.
Галактоземия у детей может протекать с более или менее выраженными нарушениями со стороны органов. Чем больше в семье заболевшего ребенка пораженных галактоземией родственников, тем более вероятность, что заболевание у него будет протекать в легкой форме. В легких случаях ребенок начинает плохо сосать грудь, отказывается от кормлений, отмечаются признаки непереносимости молочных продуктов. При более тяжелых формах признаки появляются практически сразу после рождения. При рождении ребенок имеет достаточно большие размеры и массу тела, как правило, не менее 5 кг. После первого же кормления грудью сразу возникает рвота, понос. В дальнейшем ребенок от кормлений отказывается. Масса тела начинает очень быстро и неуклонно снижаться. Печень ребенка увеличена в размерах, в ранние сроки развивается выраженная желтуха. Может отмечаться незначительное увеличение селезенки. В животе скапливается жидкость, расширяются подкожные вены. В дальнейшем развивается помутнение хрусталика глаза, нарушается зрение ребенка. Наблюдаются различные изменения показателей в лабораторных анализах. Очень информативным, однако далеко не всегда безопасным исследованием является нагрузка на обменные процессы ребенка в виде молочной пищи. После приема молока должно выявляться стойкое понижение содержания глюкозы в крови. Очень важным исследованием является определение содержания галактозы в крови, которое при заболевании очень резко повышается.
Лечение галактоземии у детей
Практически единственным мероприятием, позволяющим не допустить развития вышеуказанных нарушений, является перевод ребенка с детства на безмолочную диету. Для детей грудного возраста в настоящее время предложено огромное количество различных смесей, не содержащих компонентов коровьего молока, в частности сахара галактозы. Более старшим детям разрешается принимать в пищу яйца, масла растительного происхождения, другие продукты без содержания молока и его компонентов. При этом нельзя за счет уменьшения молока допускать снижение общего содержания калорий в пище, в этом смысле питание ребенка должно быть полноценным. Прикорм у таких младенцев вводится раньше, чем у здоровых сверстников. Смесь при этом должна быть как можно быстрее заменена другими продуктами. Каши нельзя готовить на молоке, они варятся на овощных и мясных отварах. До трехлетнего возраста молочные продукты строго запрещаются.
На данный момент пытаются разрабатывать различные медикаментозные препараты, действие которых направлено на коррекцию обмена сахаров в организме. Имеются данные об успешном применении мужских половых гормонов, но, как и всякие гормональные препараты, у детей они должны применяться с крайней осторожностью.
Дополнительными средствами для лечения галактоземии являются препараты, улучшающие работу головного мозга, влияющие на кровоток в органах, защищающие печень, улучшающие общие обменные процессы. Помутнение хрусталика при значительном нарушении зрения требует хирургического лечения.
Гликогенозы
Это группа наследственных заболеваний, связанных с нарушением утилизации в организме продукта обмена Сахаров -- гликогена. В результате данное вещество накапливается в различных органах, нарушая их функцию. Заболевание по своему течению очень многолико -- насчитывают 12 его форм. Встречается достаточно редко. Мальчики и девочки заболевают с одинаковой частотой. Ниже рассмотрены наиболее часто встречающиеся типы гликогенозов.
Первый тип заболевания -- болезнь Гирке. Назван так по имени открывшего его ученого. Нерасщепленные молекулы гликогена в основном накапливаются в печени, в меньшей степени -- в почках. В итоге данные органы значительно увеличиваются в своих размерах. При проведении анализа крови содержание глюкозы в ней снижено. Такие нарушения обмена веществ приводят к увеличению количества жиров в крови, увеличению их отложения под кожей.
Показатели глюкозы в общем анализе крови снижены, но они резко возрастают после приема пищи. При исследовании функций печени они не нарушены.
Заболевание может протекать различно. Часть больных детей очень рано погибает в результате присоединения сопутствующих заболеваний. Ведь иммунные силы организма у таких детей значительно нарушены. После наступления полового созревания прогноз становится намного более благоприятным.
Второй тип гликогеноза -- болезнь Помпе. При этом типе заболевания количество пораженных органов значительно больше, чем при предыдущем. Гликоген в больших количествах откладывается не только в печени и почках, но и в сердце, головном мозге, скелетных мышцах.
Первые признаки появляются после рождения. Ребенок категорически отказывается от кормлений, он вялый, сила мышц слабая, постоянно отмечается нарушение дыхания, кожные покровы имеют синюшный оттенок. Сердце увеличивается в размерах и принимает форму шара, что очень хорошо выявляется во время ультразвукового обследования. Появляются признаки нарушения работы сердца, при его выслушивании определяются побочные шумы. В дыхательных мышцах гликоген также накапливается в больших количествах, что приводит к нарушению нормального расправления легких во время вдоха. В итоге в них застаивается мокрота, которая, являясь хорошей средой для жизни микроорганизмов, может в дальнейшем приводить к развитию воспаления легких. В результате поражения мышц глотки может нарушаться глотание. По своему внешнему виду больные дети часто напоминают детей с врожденным синдромом Дауна. Лицо их большое, округлой формы, на нем располагаются отеки, язык большой и может не помещаться во рту, сила мышц ослаблена. Отмечается отставание в физическом и интеллектуальном развитии. Могут иметься признаки расстройств со стороны нервной системы.
При проведении биохимических анализов крови и мочи каких-либо патологических изменений не обнаруживается, так как большая часть гликогена при данной форме заболевания находится не в крови больного, а во внутренних органах.
Данный тип заболевания протекает чрезвычайно тяжело и приводит к смерти ребенка к концу первого года жизни.
Третий тип -- болезнь Форбса--Кори. Гликоген в организме больного ребенка утилизируется, но не полностью, образуются другие побочные продукты его распада, которые в конечном итоге и приводят к патологическим изменениям.
По своему течению заболевание очень сильно напоминает гликогеноз первого типа. На первый план выступает слабость скелетных мышц, вялость и утомляемость ребенка.
Прогноз более благоприятен по сравнению с первыми двумя формами и зависит от наличия поражения сердца.
Другие формы заболевания встречаются намного реже вышеописанных.
Фруктоземия
Фруктоземия встречается крайне редко. Имеет наследственную природу и связано с нарушением расщепления молекул некоторых сахаров, содержащихся в фруктах. Основными поражаемыми органами являются печень и почки. Нередко они даже полностью разрушаются.
Несмотря на то что ребенок болен с рождения, первые признаки заболевания появляются только после введения в рацион фруктов. Чаще всего они возникают после того, как ребенку первый раз в жизни дается фруктовый сок. После этого возникает рвота, иногда понос, ребенок отказывается от дальнейшего приема пищи. Если вызвавшие такую реакцию продукты не исключены из рациона и ребенок продолжает их получать, то он начинает терять в весе. В дальнейшем ребенок становится вялым, сонливым, часто падает в обморок, развиваются судороги, во время которых ребенок сильно потеет. Все эти проявления обусловлены низким содержанием в крови глюкозы. При дальнейшем прогрессировании заболевания увеличивается в размерах печень, появляется желтушность кожи, признаки нарушения работы почек. Значительно позже и реже возникают нарушения со стороны нервной системы. Зачастую родители сами замечают, какие продукты приводят к ухудшению состояния ребенка и исключают их из его рациона. Поэтому тяжелые последствия заболевания возникают достаточно редко.
Основными методами, при помощи которых можно правильно поставить диагноз в больнице, являются различные лабораторные исследования.
Прогноз всегда хороший, но лишь при условии строгого соблюдения диеты. галактоземия фруктоземия углевод лечение
Большое значение для выявления наследственных болезней обмена углеводов имеют методы качественного и количественного определения углеводов в биологических жидкостях организма, основной из которых является кровь. С целью быстрого определения содержания глюкозы в моче используются специальные индикаторные полоски бумаги. Однако данный метод является только лишь относительно точным в связи с тем, что он определяет только наличие глюкозы и ее приблизительное количество. Эти данные получают при изменении цвета индикаторной полоски после помещения ее в мочу исследуемого.
Для выявления болезней депонирования гликогена большое значение имеет нагрузочный тест. В этом случае исследуемому предлагается выпить раствор галактозы с определенным ее количеством, после чего определяют содержание глюкозы в крови. В ряде случаев для уточнения типа болезни необходимо изучение активности ферментов, принимающих участие в обмене глюкозы. Данные ферменты содержатся в мышцах, печени и некоторых других органах. Исследование проводится на кусочках ткани органа, которые берутся при проведении специального метода под названием биопсия.
Подобные документы
Основные органы, ткани и клетки, в которых найден дефект ферментов катализирующих процессы распада или синтеза гликогена. Клинические картины галактоземии, мукополисахаридозов, дисахаридной недостаточности. Лечение наследственных генетических заболеваний.
презентация [972,1 K], добавлен 15.04.2014
Массовое обследование новорожденных детей на наследственные заболевания, которое проводится в родильных домах. Раннее выявление фенилкетонури, врожденного гипотериоза, адреногенитального синдрома, галактоземии, муковисцитоза и их своевременное лечение.
презентация [1,9 M], добавлен 10.11.2014
Углеводы, их роль в биологических процессах живых организмов и человека. Характерные признаки фруктоземии. Мальтазная и изомальтазная недостаточность. Болезни, связанные с нарушением выработки ферментов. Наследственная непереносимость фруктозы, лактозы.
презентация [13,3 M], добавлен 03.12.2014
Нарушение расщепления и всасывания углеводов. Врожденная недостаточность лактазы. Основные типы регуляции углеводного обмена. Этиопатогенез, основные причины и признаки сахарного диабета, хронические осложнения. Гипергликемические состояния у человека.
лекция [24,7 K], добавлен 13.04.2009
Структура и функции генов. История расшифровки механизма развития болезней с наследственным предрасположением. Понятие, сущность и причины мутаций. Характеристика хромосомных болезней и болезней нарушения обмена веществ (аминокислот, жиров и углеводов).
К наследственным нарушениям обмена углеводов относятся различные патологические состояния, обусловленные неспособностью организма усваивать углеводы или катаболизировать их. Эти формы патологии обмена часто проявляются в раннем возрасте. Патогенез поражения нервной системы при наследственных заболеваниях обмена углеводов связан с развитием частых гипер- и гипогликемических состояний, нарушением электролитного и водного баланса, с образованием токсических продуктов метаболизма (кетокислот и др.), вызывающих метаболические и структурные нарушения в ткани мозга, с дегенерацией клеток вследствие накопления в них некатаболизируемых углеводов.
Учитывая большие возможности диетотерапии и возрастное становление ферментных систем, профилактика неврологических нарушений тесно связана с вопросами раннего диагноза.
Диспансерное наблюдение за больными и успехи в организации специализированной помощи позволили реально решить вопрос о доклинической диагностике, рассматривая каждого ребенка, рожденного в семье, где есть патология углеводного обмена, как потенциального больного. Гетерозиготные носители патологического гена выявляются с помощью нагрузочных тестов и исследования активности ферментов. Эти данные используются при решении вопросов медико-генетического консультирования.
Среди заболеваний, обусловленных нарушением обмена углеводов, выделяют состояния, проявляющиеся непереносимостью того или иного углевода, входящего в состав продуктов питания, и заболевания, обусловленные нарушением метаболизма гликогена.
Галактоземия
Наследственная непереносимость галактозы описана К. Reuss в 1908 г., наследуется аутосомно-рецессивно, частота в популяции — 1 : 70 000, частота гетерозиготных носителей — 1 : 268.
Патология обусловлена резким снижением активности фермента галактозо-1-фосфат уридилтрансферазы (ГФУТФ), что приводит к накоплению в крови и тканях галактозы и галактозо-1-фосфата. В патогенезе важное место принадлежит формированию гипогликемического синдрома, ацидоза, гипокалиемии.
Патоморфологические изменения при галактоземии наиболее типичны в печени. Они характеризуются жировой дистрофией, околодольчатым некрозом, циррозом. Выявляется также отек и набухание мозга, вздутие ядер олигодендроглии в больших полушариях мозжечка.
Первые клинические симптомы непереносимости галактозы появляются вскоре после рождения, как только ребенок начинает получать молоко. При полном отсутствии ГФУТФ у новорожденного возникают неукротимая рвота, желтуха, быстро нарастает гипотрофия, развиваются гепатомегалия, асцит. Прогрессирующие диспептические нарушения сопровождаются симптомами нейротоксикоза, которые проявляются судорожным синдромом, нистагмом, грубыми глазодвигательными нарушениями (плавающие движения глазных яблок), затруднением сосания и глотания.
Синдром мышечной гипотонии различной выраженности сочетается с арефлексией. Снижены или отсутствуют рефлексы новорожденного, особенно рефлекс Моро, хватательный, группа защитных рефлексов. Нарастает отек мозга и легких, летальный исход наступает через несколько месяцев.
При частичном снижении активности ГФУТФ течение заболевания может быть ремиттирующим. Патология проявляется в первые месяцы жизни, ее дебют обусловлен расширением молочной диеты. Характерны эпизоды рвоты, показатели роста и массы ниже нормы, отмечаются субиктеричность кожных покровов, легкое или умеренное увеличение печени, характерны формирующиеся катаракты (чаще двусторонние). Патология черепномозговых нервов вариабельна: сходящееся косоглазие, нистагм. Ребенок отстает в двигательном развитии, однако наличие патологических поз и установок не характерно. Синдром мышечной дистонии нередко сменяется гипотонией со снижением сухожильных рефлексов. Лицо гипомимично. Преобладает отрицательный эмоциональный комплекс. Снижение зрения приводит к запаздыванию формирования ориентировочно-познавательных реакций. Отмечается задержка психического развития. Эпизоды судорожных пароксизмов делают еще более сомнительным прогноз психического развития.
При биохимическом обследовании больных галактоземией обнаруживаются галактозурия, протеинурия, гипераминоацидурия (тотальная); повышение содержания галактозы в крови при гипогликемии. Характерны патологическая кривая при нагрузке галактозой, ацидоз. Диагноз подтверждается хроматографической идентификацией галактозы, накоплением в эритроцитах галакто-зо-1-фосфата и снижением или отсутствием активности фермента галактозо-1-фосфат-уридилтрансферазы.
Дифференциальный диагноз проводят со всеми меллитуриями (избыточным выделением углеводов с мочой), в том числе и сахарным диабетом, с гипогликемией новорожденных, гепатитами, цистинозом, синдромами гипераминоацидурий.
Лечение диетическое. Из рациона исключается молоко и продукты, содержащие галактозу. Детям раннего возраста рекомендуется пища без галактозы: смесь яиц с сахаром, маргарин, рисовая мука, белки животного происхождения, обогащенные экстрактами овощей, мяса, витаминами. В тяжелых случаях показана симптоматическая терапия, направленная на уменьшение ацидоза, противосудорожная терапия, обменное переливание крови. С возрастом непереносимость галактозы снижается, что обусловлено повышением активности УДФ-Гал-пирофосфорилазы, способствующей превращению галактозы побочным путем. Активирующим действием на УДФ-Гал-пирофосфорилазу обладают прогестерон и тестостерон. При ранней диетической коррекции физическое и психическое развитие детей нормальное.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Галактоземия (ГАЛ) – группа наследственных нарушений обмена углеводов, при котором в организме накапливается избыток галактозы и ее метаболитов, что обусловливает клиническую картину заболевания и формирование отсроченных осложнений. Тип наследования всех форм галактоземии - аутосомно-рецессивный.
Особенности кодирования заболевания или состояния (группы заболеваний или состояний) по Международной статистической классификации болезней и проблем, связанных со здоровьем
E74.2 - Нарушения обмена галактозы (Недостаточность галактокиназы. Галактоземия)

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
Классификация
В зависимости от дефекта одного из трех основных ферментов, участвующих в обмене галактозы выделяют три типа ГАЛ:
I. Классический - галактоземия I типа, обусловленная дефицитом фермента галактозо-1-фосфат-уридилтрансферазы (ГАЛТ) и наличием гомозиготных или компаунд-гетерозиготных мутаций в гене GALT (G/G). Отдельно выделяют вариант Дуарте (D/D) и галактоземию- Дуарте (G/D), при которых полиморфный вариант находится в гомозиготном состоянии или компаунд-гетерозиготном состоянии с патогенной мутацией.
Этиология и патогенез
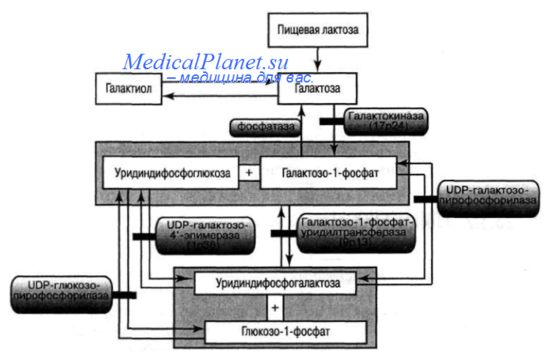
Галактоза (от греческого слова galaktos - молоко) - моносахарид из группы гексоз, изомер глюкозы. Несмотря на большое сходство молекул глюкозы и галактозы, превращение последней в глюкозу требует нескольких ферментативных реакций, которые протекают в цитоплазме клетки (рис. 1). Галактоза имеет важнейшее значение для роста и развития организма и является компонентом грудного молока.
Галактоза не только является значимым источником энергии для клетки, она играет важную пребиотическую роль, служит необходимым пластическим материалом для образования гликопротеидов, гликолипидов и других сложных соединений, используемых организмом для формирования клеточных мембран, нервной ткани, процессов миелинизации нейронов и др. Нарушение метаболизма галактозы, наблюдаемое при галактоземии, неизбежно приводит к расстройству функционирования многих органов и систем организма.
Большое количество потребляемых в течение дня пищевых продуктов (в первую очередь молочные продукты) содержат лактозу, из которой в кишечнике в результате гидролиза образуется галактоза; некоторые продукты питания содержат галактозу в чистом виде. У человека галактоза может образовываться эндогенным путем, подавляющее ее количество синтезируется в процессе ферментативных реакций между уридиндифосфатглюкозой (УДФ-глюкозой) и УДФ-галактозой, а также в процессе обмена гликопротеинов и гликолипидов.
Галактоземия относится к наследственным болезням углеводного обмена и объединяет несколько генетических форм заболеваний. Галактоземия тип I (ГАЛ I) обусловлена мутациями в гене GALT, картированном на 9p13.3, что приводит к дефициту галактозо-1-фосфатуридилтрансферазы (ГАЛТ). Галактоземия тип II (ГАЛ II) обусловлена мутациями в гене GALK1, картированном на 17q25.1, что приводит к дефициту галактокиназы (ГАЛК). Галактоземия тип III (ГАЛ III) обусловлена мутациями в гене GALE, картированном на 1р36.11, что приводит к дефициту уридин-дифосфат (УДФ)-галактозо-4-эпимеразы (ГАЛЭ) (рис 1).
В результате недостаточности любого из трех ферментов – ГАЛТ, ГАЛК или ГАЛЭ – в крови повышается концентрация галактозы. При снижении активности ферментов ГАЛТ и ГАЛЭ, помимо избытка галактозы, в организме пациента накапливается также избыточное количество галактозо-1-фосфата, что на сегодняшний день считается основным патогенетическим фактором, обусловливающим большинство клинических проявлений ГАЛ и формирование отсроченных осложнений. Избыток галактозы в организме может метаболизироваться в других биохимических путях: она может превращаться в галактитол. Накопление галактитола в крови и тканях и повышение его экскреции с мочой наблюдается при всех формах ГАЛ; в хрусталике глаза избыток галактитола способствует формированию катаракты. Имеются сведения о том, что высокое содержание галактитола в тканях мозга способствует набуханию нервных клеток и формированию псевдоопухоли мозга у отдельных пациентов.
Патологические процессы при ГАЛ обусловлены не только токсическим действием указанных продуктов, но и их тормозящим влиянием на активность других ферментов, участвующих в углеводном обмене (фосфоглюкомутазы, глюкозо-6-фосфатдегидрогеназы), следствием чего является гипогликемический синдром. Предполагается также, что предрасположенность к сепсису у новорожденных с ГАЛ тип I обусловлена ингибированием бактерицидной активности лейкоцитов.
Галактоземии II типа (ГАЛ II) обусловлена мутациями в гене GALK1, картированном на 17q25.1. Описано около 50 мутаций, частых мутаций не описано. Известна одна мутация, p.P28T, которая часто встречается у цыган [17].
Галактоземия тип III (ГАЛ III) обусловлена мутациями в гене GALE, картированном на 1р36.11. Одна из самых редких форм ГАЛ. Известно около 30 мутаций, частых мутаций не описано.
Читайте также: