
Лизосомные болезни накопления реферат
Обновлено: 03.07.2024
Лизосомальные ферменты разрушают макромолекулы либо самой клетки (например, когда перерабатываются структурные компоненты клетки), либо захваченные извне. Унаследованные дефекты или недостатки лизосомальных ферментов (или других лизосомальных компонентов) могут привести к накоплению недеградированных метаболитов. По наличию многочисленных конкретных недостатков болезни накопления обычно группируют биохимически в зависимости от накапливаемого метаболита. Подгруппы включают
Наиболее важными являются мукополисахаридоз и сфинголипидоз. Тип 2 гликогеноза является расстройством лизосомального накопления, но большинство гликогенозов – нет.
Поскольку ретикулоэндотелиальная клетка (например, в селезенке) богата лизосомами, ретикулоэндотелиальная ткань участвует в ряде лизосомных болезней накопления, но, как правило, затронуты ткани, наиболее богатые субстратами. Таким образом, мозг, который богат ганглиозидами, особенно страдает от ганглиозидозов, в то время как мукополисахаридозы поражают многие ткани, так как мукополисахариды присутствуют по всему телу.
Мукополисахаридозы (МПС)
Возраст возникновения, клинические проявления и тяжесть варьируют в зависимости от типа (см. таблицу Мукополисахаридоз Мукополисахаридоз Лизосомальные ферменты разрушают макромолекулы либо самой клетки (например, когда перерабатываются структурные компоненты клетки), либо захваченные извне. Унаследованные дефекты или недостатки. Прочитайте дополнительные сведения ). Общие проявления включают грубые черты лица, задержку нервного развития и регресс, контрактуры суставов, органомегалию, жесткие волосы, прогрессирующую дыхательную недостаточность (в связи с обструкцией дыхательных путей и ночным апноэ), сердечные пороки, скелетные изменения и подвывих шейных позвонков.
Мукополисахаридоз диагностируется на основании анамнеза болезни, данных осмотра, наличия костных аномалий (например, множественных дизостозов), обнаруженных при исследовании скелета, и повышения общего и фракционированного уровня гликозаминогликанов. Диагноз подтверждают на основании анализа ДНК и/или анализа ферментов в культуре фибробластов (пренатально) или периферических лейкоцитов (после рождения). (Также см. Исследования при подозрении наследственных нарушений обмена веществ Начальное тестирование Большинство наследственных нарушений метаболизма (врожденные нарушения обмена веществ) являются редкими, и, следовательно, для их диагностики необходим более высокий индекс подозрения. Своевременная. Прочитайте дополнительные сведения ). Дополнительное тестирование проводят для контроля орган-специфических изменений (например, эхокардиография для пороков клапанов, аудиометрия для выявления изменений слуха).
Лечение мукополисахаридоза I типа состоит в восполнении фермента ларонидазы, которая эффективно останавливает прогрессирование заболевания и все его осложнения, не связанные с центральной нервной системой. Также используется трансплантация гемопоэтических стволовых клеток (ГСК). Сочетание восполнения ферментов и трансплантации гемопоэтических стволовых клеток исследуется. Для пациентов с MPS типа IV-A (синдром Моркиа), возмешение фермента элосульфазой альфа может улучшить функциональный статус, в том числе подвижность.
1 Тюменский государственный медицинский университет Министерства здравоохранения Российской федерации
Лизосомные болезни накопления (LSDs – англ. Lysosomal storage diseases) – группа редко встречающихся заболеваний накопления, являющихся следствием первичной генной мутации. Известно уже более 50 разновидностей LSDs, и традиционно они классифицируются согласно химическим свойствам накопленного комплекса. Отдельные случаи лизосомных болезней встречается не чаще, чем у одного из 100 тысяч ново-рождённых, при этом распространённость конкретного заболевания в различных популяциях может значительно варьироваться. В статье представлен обзор научных публикаций, в которых рассматриваются современные стратегии и принципы лечения лизосомных болезней накопления. Рассматриваются положительные и отрицательные моменты применения различных методик, среди которых выделяются: фермент-заместительная терапия, транс-плантация стволовых гемопоэтических клеток, использование молекул фармакологических шаперонов и генотерапии. Проанализированы проблемы, возникающие при использовании данных терапевтических стратегий.

1. Бучинская, Н. В. Современные подходы к терапии мукополисахаридозов у детей, / Н.В. Бучинская и др. // Вопросы современной педиатрии. – 2014. – №3. – Том 13. – С. 35-43.
2. Бушуева Т.В. Современный взгляд на проблему фенилкетонурии у детей: диагности-ка, клиника, лечение. / Т.В. Бушуева // Вопросы современной педиатрии. – 2010. - №1. – Том 9. – С. 157-159.
3. Гасина, А.А., Гусина, Н.Б. Современные подходы к лечению лизосомных болезней накопления, /А.А. Гасина, Н.Б. Гусина// Медицина. –2009.– № 3. – С. 27-32.
4. Захарова Е. Современные подходы к лечению болезни Гоше и других лизосомных болезней накопления. / Е. Захарова и др. // RASUS. Редкие болезни в России. – 2014. - №2. – С. 16-18.
5. Захарова, Е. Ю. Лизосомные болезни накопления, / Е.Ю. Захарова // Педиатрия и дет-ская хирургия. – 2010. - №4. – С. 49-53.
6. Цыгин, А.Н. Нефропатический цистиноз, / А.Н. Цыган и др. // Клиническая нефроло-гия. – 2011. – №4. – С. 20-23.
9. Cheng SH, Smith AE. Gene therapy progress and prospects: gene therapy of lysosomal stor-age disorders // Gene Therapy. – 2003. - №10. – Р.1275-1281.
10. Fan JQ, Ishii S. Active site-specific chaperone therapy for Fabry disease. Yin and Yang of enzyme inhibitors // FEBS J. – 2007, 274: 4962-4971.
11. Grubb JH, Vogler C, Levy B, Galvin N, Tan Y, Sly WS. Chemically modified beta-glucuronidase crosses blood-brain barrier and clears neuronal storage in murine mucopoly-saccharidosis VII // Proc Natl Acad Sci USA. – 2008. №105. – Р. 2616-2621
12. Marca G. Lysosomals. Physician's Guide to the Diagnosis, Treatment, and Follow-Up of In-herited Metabolic Diseases // Springer Berlin Heidelberg. - 2014. — P. 785-793.
13. Parenti G, Andria G, Valenzano KJ. Pharmacological Chaperone Therapy: Preclinical De-velopment, Clinical Translation, and Prospects for the Treatment of Lysosomal Storage Dis-orders // Molecular Medicin. – 2015. №23. – Р. 113-1148.
15. Sands M.S., Davidson B.L. Gene Therapy for Lysosomal Storage Diseases // Molecular Therapy. – 2006. №13. – Р.839-849.
16. Scriver, Charles R.: The Metabolic & Molecular Bases of Inherited Disease // Scriver, R. Charles. - 8th ed. New York: McGraw-Hill., 2001. – P. 6338.
Введение
Лизосомные болезни накопления (LSDs – англ. Lysosomal storage diseases) – группа редко встречающихся наследственных заболеваний, вызываемых генетически обусловленным дефектом лизосомных гидролитических ферментов, клеточных транспортеров и белков, входящих в состав мембран. В конечном счете это приводит к накоплению макромолекул и метаболитов в клетках, а как следствие к повреждению отбельных тканей, органов и систем организма. Болезни накопления являются следствием первичной генной мутации.
Известно уже более 50 разновидностей LSDs, и традиционно они классифицируются согласно химическим свойствам накопленного комплекса. Отдельные случаи LSDs встречается не чаще, чем у одного из 100 тысяч новорождённых, при этом распространённость конкретного заболевания в различных популяциях может значительно варьироваться [12].
Большинство LSD наследуются аутосомно-рецессивно, за исключением заболеваний, которые наследуются сцеплено с X хромосомой. К ним относятся мукополисахаридоз Хантера (МПС II) и болезнь Фабри, являющиесяся рецессивными Х-сцепленными заболеваниями, а также синдром Данона, являющийся доминантным Х-сцепленным заболеванием.
Заболевания характеризуются тяжестью последствий с точки зрения здоровья пациентов, социальных и экономических затрат. LSD затрагивают многие ткани и органы, поэтому для них характерна множественность внутренних, соматических и неврологических проявлений. Среди клинических проявлений, связанных с центральной нервной системой, чаще можно встретить прогрессирующие нейродегенеративные заболевания и умственную отсталость [2, 5].
Мультиполярнось фенотипических проявлений LSD и их патофизиология представляют определенную сложность для терапевтических вмешательств. Способы лечения должны быть направлены как на устранение причины заболевания, на восстановление пораженных органов, так и на повышение качества жизни пациентов. Начиная с конца 20 века было внедрено много терапевтических методов, однако у данных подходов есть ограничения, и важные проблемы остаются нерешенными. Кроме того, у каждого из доступных подходов есть строгие признаки и критерии применимости для отдельного LSD, или даже для отдельных пациентов. В принципе, ни у одного из вариантов нет возможности устраняя
ить все заболевания, поэтому в текущих условиях все силы направлены на поиски альтернативных терапевтических подходов для лечения LSDs на основе инновационных стратегий [3, 14].
Существует три основные научные стратегии в лечении LSD, которые ориентированы на снижение концентрации патологического субстрата, восстановление и поддержание конформации поврежденного белка, либо на устранение первопричины – генетической мутации.
- Воздействие на патологический субстрат
Стратегия воздействия на патологический субстрат заключается в двух подходах: химическая модификация продукта накопления и снижение синтеза субстрата. Модификация продукта применяется в том случае, если происходит повреждение транспортных систем лизосом [3, 6]. Химическое превращение накопившегося субстрата позволяет получить такое вещество, которое может без трудностей выходить из лизосом через их мембранные переносчики. Данный механизм лежит в основе лечения цистиноза. Цистиноз - редкое аутосомно-рецессивное заболевание, причиной которого является мутация в гене CTNS, кодирующем лизосомальный переносчик цистина. При этом заболевании в различных органах и тканях происходит накопление цистина в лизосомах и отложение его кристаллов. Для лечения данной поталогии используется цистеамин – единственное вещество, являющееся супрессором отложения цистина внутри лизосом. Препарат проникает в органеллу, где расщепляет цистин на две молекулы цистеина и затем соединяется с одной из них с помощью дисульфидной связи. Образующиеся при этом цистеин-цистеаминовый комплекс и цистеин не нуждаются в цистинозине для выхода из лизосомы. Дисульфидное соединение, являющееся аналогом лизина, переносится через лизиновый транспортер [6].
Большинство известных на данный момент групп LSDs связаны со сбоем определенных стадий катаболизма (мукополисахаридозы, гликосфинголипидозы и т.д.), и их патогенетические механизмы обусловлены накоплением в клетках нерасщепленного субстрата [16]. Методика снижения синтеза субстрата применяется в том случае, если сохраняется малая активность дефектного фермента. При субстратредуцирующей терапии происходит снижение катализируемого вещества, этим путем достигается равновесие между синтезом субстрата и его диссимиляцией. Об успешности данного метода заговорили, когда были получены положительные результаты на животных моделях болезни Тея–Сакса и Сандхоффа. Использование данной методики приводило к снижению концентрации субстрата в центральной нервной системе и, как следствие, к уменьшению степени выраженности симптомов заболевания. В последующем подобные эксперименты проводили на животных моделях болезни Гоше типа I, где также был достигнут положительный эффект [1, 5].
- Коррекция функций измененного фермента
Большинство терапевтических подходов при лечении LSDs направлено на восстановление потерь измененного энзима путем увеличения его содержания в клетках и тканях.
К данным методикам относится фермент-заместительная терапия (ФЗТ), которая в прошлом являлась огромным рывком в лечении LSDs, а на данный момент является стандартом коррекции многих заболеваний (болезнь Гоше, Фабри, Помпе и мукополисахаридозы). Обоснования эффективности ФЗТ развивались с момента обнаружения молекулярных механизмов, которые участвуют в сортировке и распределении синтезируемых ферментов. ФЗТ основывается на концепции, согласно которой лизосомные гидролазы могут усваиваться клетками и тканями через маннозный или манноза-6-фосфатный транспортер, затем проникают в лизосомы, где восполняют функцию дефектного энзима [8]. Еще одним толчком для эволюции заместительной терапии стало развитие технологий, позволяющих производить очистку и разнообразные манипуляции с энзимами для их нацеленной доставки в клетки и ткани.
Несмотря на успехи применения, ФЗТ имеет определенные ограничения. Основные трудности заключаются в распределении ферментов, так как белковые молекулы имеют большие размеры и не могут свободно проходить через биологическую мембрану. В большинстве случаев рекомбинантные энзимы не могут проходить через гематоэнцефалический барьер (ГЭБ). В связи с этим терапия не оказывает влияние на неврологические проявления заболевания. Здесь как альтернативный способ доставки препаратов в центральную нервную систему может быть применен интратектальный путь введения. В качестве другого метода преодоления ГЭБ были предприняты попытки химической модификации b-глюкуронидазы (при дефекте этого фермента развивается МПС VII типа). Измененные молекулы отменяли маннозный и маннозо-6 фосфатный путь поглощения фермента, в результате происходило накопление фермента в плазме крови и увеличение его доставки к клеткам головного мозга через неизвестный путь [11].
Также существует проблема целенаправленной доставки фермента. Так при болезни Помпе наблюдается метаболическая миопатия в результате дефицита кислой α-1,4-глюкозидазы. Действие лечебного фермента направлено на коррекцию деятельности основных пунктов потребления – сердце и мышцы, но это представляет определенную сложность, так как большая часть энзима будет захвачена печенью и только незначительная часть препарата доставляется в мышцы. На эффективность ФЗТ также могут влиять аспекты патофизиологии и вторичные клеточные нарушения, вызванные накоплением субстрата. К тому же побочным эффектом введения рекомбинантных ферментов ослабленным пациентам может служить иммунный ответ. Следует отметить и то, что для поддержания устойчивого уровня корректирующих ферментов необходимо постоянное проведение инъекции или установка имплантируемой венозной порт-системы. Не стоит игнорировать и экономический аспект вопроса. Лечение одного больного может обойтись в несколько сотен тысяч долларов [7].
Применение ФЗТ основано на экзогенном введении препаратов, но применяются и методы, направленные на эндогенное замещение энзимов путем пересадки стволовых клеток – трансплантация костного мозга. Практически для всех LSDs был опробован данный терапевтический метод, но большинство экспериментов касаются лишь единичных случаев и, в большинстве случаев, это не приводило к коррекции всех осложнений заболевания. Первое заболевание, для которого была проведена ТКМ, и достигнуты наилучшие результаты – МПС I типа [5, 14]. Эффективность лечения зависит от сроков начала процедуры и является продуктивным до 2 лет, когда поражение центральной нервной системы не столь существенно. После пересадки гемопоэтических клеток у пациентов наблюдается восстановление собственной активности ферментов до нормальных значений и снижение уровня экскреции глюкозаминоглюканов, до уровня верхней границы нормы для данного возраста [1]. Существуют и проблемы в применении данного метода. Они заключаются в высоком риске данной манипуляции, в сложности подбора донора и в посттрансплантационных осложнениях, однако ТКМ остается на сегодняшний день стандартным методом лечения МПС I типа.
Для лечения других LSDs роль трансплантации гемопоэтических клеток менее доказана или противопоказана пациентам с тяжелыми нейродегенеративными формами. В перспективе рассматривается применение мезенхимальных и других стволовых клеток костного мозга, а также сочетание ТКМ с другими методами лечения [5, 14].
В качестве фармакологических шаперонов было предложено использование низкомолекулярных, чаще гидрофобных, молекул, стабилизирующих нативную конформацию специфических для них белков. В экспериментах in vitro было обнаружено, что некоторые соединения способны связываться с каталитическим центром энзима и поддерживать его структуру. Работа над культурами клеток с мутациями в гене α-галактозидазы (болезнь Фабри) показала, что при введении в культурную среду 1-deoxygalactonojirimycin в концентрациях близких к ингибиторным происходит стабилизация измененного фермента. Введение вещества трансгенным мышам с паталогией гена α-галактозидазы приводит к повышению активности энзима в сердце, почках, селезенке и других тканях [5, 10]. Данные результаты говорят нам о том, что конкурентные ингибиторы восстанавливают конформацию белка и обеспечивают его доставку в другие структуры клетки. Подобные наблюдения позволяют применять методику фармакологических шаперонов для многих заболеваний, связанных с мисенс-мутациями.
Целью генотерапии является доставка нормальной ДНК в клетки пациента и синтез утерянного фермента. В этих целях в лабораториях применяют различные вирусные векторы (ретровирусы, аденовирусы, аденоассоциированные вирусы) и другие методики.
В основном исследование по применению вирусных векторов проводятся на животных моделях и дают положительные результаты. Так огромный потенциал показывает система переноса генов с применением аденоассоциированных вирусов (AAV). Впервые разработан и применен пожизненно был AAV 2 типа. Внутримышечные инъекции данного вектора проводились трансгенным мышам с моделью болезни Помпе, Фабри и МПС VII типа. В результате наблюдался высокий уровень активности дефектного фермента в инъецированной мышце, но относительно низкий или неопределяемый уровень в системе крови. Инъекции AAV-2 в паренхиму печени и мышцы взрослым мышам с МПС VII типа приводят к относительно высокому уровню экспрессии в тканях и снижению количества накопившегося субстрата. Однако такой подход обеспечивает лишь незначительные клинические улучшения [9, 15].
О значительных улучшениях при внутривенном введении вектора свидетельствуют биохимические, гистологические и клинические улучшения в нескольких мышиных моделях LSDs. Положительные изменения и сокращение накопившегося метаболита наблюдаются во многих тканях после внутривенного введения вектора AAV-2 у взрослых животных с МПС VII, болезнью Помпе и болезнью Фабри. Еще более впечатляющие результаты, такие как улучшение роста костей и развития скелета, нормальное функционирование органов зрения и слуха, наблюдаются у мышей с МПС VII после внутривенного введения вектора в неонатальный период. Исследования новорожденных доказали полезность раннего вмешательства для профилактики прогрессирующего заболевания [9, 15]. Гепотерапия является перспективным направлением в лечении LSDs, но существуют проблемы, которые еще предстоит решить в будущем.
Выводы:
Лизосомные болезни накопления – это обширная группа заполеваний, причинами которых являются мельчайшие сбои в молекулярной физиологии клеток. Существуют различные способы борьбы с данными дефектами, однако каждый из них имеет свои преимущества и недостатки. Современные технологи позволяют ученым все глубже познавать механизмы развития заболеваний и помогают им приблизиться к способам устранения дефектов. Различные методики (генотерапия, использование фармакологических шаперонов) являются будущим медицины и вскоре могут стать основными способами лечения лизосомных болезней накопления.
Липоидозы – наследственные заболевания, связанные с нарушением метаболизма жиров, отложением липидов и их метаболитов в различных органах и тканях. Общие клинические проявления представлены прогрессирующим расстройством интеллектуальных и двигательных функций, поражением костей, кожи, центральной нервной системы, глаз и внутренних органов (печени, почек, селезенки). Диагностика основана на лабораторных исследованиях ферментативной активности, количества токсичного субстрата, наличия мутации в генах. Лечение включает ферментозаместительную, субстратредуцирующую и симптоматическую терапию.
МКБ-10
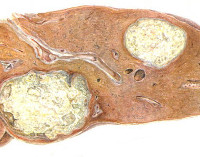
Общие сведения
Синонимичные названия липоидозов – липидозы, ретикулоэндотелиозы, лизосомные болезни накопления липидов. К данной группе относится сфингомиелиноз, болезнь Тея-Сакса, болезнь Гоше, семейные гиперлипидемии и некоторые другие заболевания. Общей характеристикой является патологическое накопление липидов внутри клеток организма в результате дефекта ферментных систем. Липоидозы относятся к группе редких (орфанных) заболеваний. Их распространенность очень низка, для отдельных типов патологий составляет от 1:40 тыс. до 1:1 млн. и реже. Суммарная частота – 1:7 тыс. Большинство липидозов имеют прогредиентное течение, приводят к инвалидизации и ранней смерти.
Причины липоидозов
Определяющим фактором развития липидозов является генетический дефект, который обуславливает полную или частичную недостаточность лизосомальных ферментов, расщепляющих сложные липиды. Наследование болезней происходит по аутосомно-рецессивному механизму. Это означает, что новорожденный оказывается больным, если получает мутационный ген от каждого родителя (имеет пару дефектных генов в аллели). Когда мутация передается только от матери или отца, то ребенок является ее носителем и остается здоровым.
Исключительный механизм передачи дефекта у болезни Фабри. В отличие от других липидозов, она наследуется по X-сцепленному рецессивному типу. Гемизиготные пациенты мужского пола больны, передают мутацию только дочерям. У девочек заболевание всегда проявляется при наличии двух рецессивных (измененных) генов. Иногда симптомы липидоза проявляются у пациенток с одним дефектным геном, когда доминантный ген оказывается инактивированным (причины этого не выяснены).
Патогенез
Патогенетической основой большинства липоидозов является генетически детерминированный ферментативный дефект – энзимопатия. Вследствие этого лизосомы клеток накапливают липиды и промежуточные продукты их метаболизма, что ведет к прогрессивному нарастанию нарушения функций органов. При синдроме Вольмана определяется недостаточность кислой этеразы, становится невозможным полный цикл обмена холестерола, повышается содержание его эфиров в лизосомах селезенки, печени, надпочечников, кишечника и костного мозга. У пациентов с болезнью Гоше снижено или полностью отсутствует производство бета-глюкозидазы; в печени, селезенке, костном мозге скапливаются продукты расщепления сфинголипидов. Болезнь Нимана-Пика развивается на базе дефицита сфингомиелазы, характеризуется повышением количества сфингомиелина во многих клетках, особенно в гепатоцитах, нейронах. При идиотии Тэя-Сакса существует дефект N-ацетилгексозаминидазы, происходит накапливание ганглиозидов в головном мозге.
Классификация
Наследственные нарушения метаболизма сложных липидов представлены группой различных по происхождению заболеваний, которые на патогенетическом уровне связаны с патологией жирового обмена. Для липоидозов характерно скопление сложных липидных соединений внутри лизосом клеток. В зависимости от того, какой липид не расщепляется до конца и откладывается в тканях, выделяют несколько типов болезней:
- Гликолипидозы. При данном типе заболеваний невозможен полный распад гликолипидов – соединений, состоящих из углеводов и жирных кислот. Гликолипидозы представлены цереброзидозами (сфинголипидоз Гоше, болезнь Краббе), сульфатидозами (метахромотическая лейкодистрофия), церамидолигозидозами (болезнь Фабри, церамидлактозидлипоидоз), ганглиозидозами (болезнь Сандхоффа, ранняя детская амавротическая идиотия).
- Липопротеинемии. Обусловлены патологией обмена липидов плазмы крови, основанной на генетическом дефекте ферментов или рецепторов клеток. Липиды плазмы – жирные кислоты, триглицериды, холестерин. Липопротеинемии включают семейную гиперхолестеринемию, комбинированную семейную гиперлипидемию и семейные гиперлипидемии типов I-V.
- Сфингомиелиноз. Синонимичное название – болезнь Ниманна-Пика. При развитии этого заболевания в ретикулоэндотелиальных клетках увеличивается содержание фосфолипидного соединения сфингомиелина.
Симптомы липоидозов
Клиническая картина липидозов определяется особенностями вовлечения в патологический процесс органов и систем. При болезни Гоше I типа поражается печень, селезенка, кости и костный мозг. Симптомы включают увеличение размеров печени, хрупкость костей, анемию, лейкопению, снижение свертываемости крови. II тип заболевания разворачивается с преимущественным повреждением ЦНС и печени. Наблюдаются судорожные приступы, мышечный гипертонус, спастичность, интеллектуальная недостаточность, расстройства акта глотания. У людей с галактозилцерамидным липидозом (болезнью Краббе) снижается функциональность миелиновой оболочки. Развивается гипервозбудимость, рвота, судороги, задержка психомоторного развития с прогрессирующим снижением интеллекта и зрения.
При метахромотической лейкодистрофии патологически изменяется миелиновая оболочка. Основные симптомы – гипотония мышц рук и ног, снижение (отсутствие) сухожильных рефлексов, атаксия, атрофия зрительных нервов, нистагм, спастический тетрапарез, глухота, интеллектуальное и моторное недоразвитие. Клинические признаки болезни Андерсона-Фабри разнообразны, одними из наиболее распространенных являются нейропатическая боль (умеренной и легкой выраженности, в ступнях и ладонях) и кризы Фабри – сильные жгучие боли в конечностях приступообразного характера. Дополнительно возникает гипогидроз или ангидроз, непереносимость физических нагрузок, ангиокератомы, расстройства слуха, сердечно-сосудистой и почечной функций.
При развитии синдрома Сандхоффа определяется общая вялость, гипотонус мышц конечностей, трудности сосания и глотания, прогрессирующая задержка моторного и психического развития, двигательная слабость, шумы в сердце, судороги, слепота, увеличение селезенки. При ранней детской амавротической идиотии больше всего поражается центральная нервная система. К концу первого полугодия ухудшается реакция детей на внешние сигналы (лица близких, игрушки), утрачиваются двигательные навыки, снижается познавательный и игровой интерес. Нарушается зрение вплоть до слепоты. Формируются судорожные припадки.
Первые симптомы сфингомиелиноза – вялость, малоподвижность, апатичность, отказ от еды, рвота. Позже увеличивается живот (гепатомегалия), конечности становятся худыми, кожа приобретает коричневатый оттенок, периоды заторможенности сменяются гипервозбуждением. Дети отстают в психофизическом развитии. Диагностируется умеренная гидроцефалия, гипертермия, спастический парез ног и рук, приступообразная асфиксия.
Проявления гиперлипопротеинемий обнаруживаются в возрасте до 10 лет. Для всех форм болезней свойственно отложение подкожного жира в виде ксантом, а также абдоминальные боли, панкреатит, гепатоспленомегалия (скопление липидов в селезенке, печени). При комбинированной семейной гиперлипидемии и гиперлипидемии II типа возможен ранний атеросклероз сосудов. При гиперлипидемиях IV и V типа – снижение чувствительности к глюкозе, ИБС.
Осложнения
Липоидозы протекают с развитием недостаточности жизненно важных органов. Наиболее частыми осложнениями становятся заболевания сердца и сосудов, ЦНС, почек, легких, печени. Пациенты страдают от атеросклероза сосудов, сердечной и дыхательной недостаточности, ишемической болезни сердца, хронической почечной и печеночной недостаточности, аденом и цирроза печени, инсультов, транзиторных ишемических атак. При типах заболеваний, совместимых с жизнью, зачастую нарушено двигательное и психическое развитие. Многие больные оказываются неспособны к самообслуживанию, обучению и овладению профессией, нуждаются в пожизненном уходе со стороны окружающих.
Диагностика
Липоидозы не являются узкоспециализированными заболеваниями, поэтому их выявлением и лечением занимаются педиатры, гематологи, гастроэнтерологи, ревматологи, неврологи, психиатры и врачи-генетики. На первичном этапе диагностики проводится сбор семейного анамнеза: при наследственном характере ферментопатии у больного могут быть родственники с подтвержденным диагнозом липидоза. При сборе клинических данных специалисты обращают внимание на время начала симптомов: чаще всего болезнь проявляется в период новорожденности или первого года жизни, редко – у детей постарше или взрослых. К специфическим методам обследования пациентов относят:
- Анализ активности дефектного фермента. Исследованию подвергаются различные биоматериалы – плазма крови, лейкоциты, сухие пятна крови, культура фибробластов кожи, биопсийный материал почек, печени. При липоидозах определяется недостаток активности определенного фермента: от легкого снижения до полного отсутствия.
- Количественное исследование липидов. В крови и биопсийном материале органов анализируется содержание патологически накапливаемых липидов и промежуточных продуктов их обмена. У больных липоидозом показатели превышают норму. Параллельно изучаются изменения строения пораженных клеток.
- Секвенирование ДНК. Выявление дефектного гена в хромосоме является наиболее точным, но трудоемким методом диагностики наследственных болезней. Широко применяется в рамках перинатальной и преимплантационной диагностики, в случаях, когда вышеперечисленные анализы не дают однозначного представления о диагнозе.
- Визуализирующие исследования органов. Дополнительно проводится диагностика состояния пораженных органов – сердца, печени, желчного пузыря, селезенки, легких, почек, головного мозга. Используется УЗИ, МРТ, КТ, ЭКГ, ЭЭГ. Процедуры позволяют оценить размеры, выявить структурные изменения и новообразования в органе.
Лечение липоидозов
Терапия данной группы заболеваний – сложная задача для врачей разных специальностей. Методы лечения несовершенны и продолжают разрабатываться, при некоторых видах болезней возможно добиться лишь временного улучшения самочувствия больного, при других достижима стойкая ремиссия. Общая схема медицинской помощи больным состоит из трех компонентов:
- Ферментозаместительная терапия. В организм пациентов вводятся препараты с искусственно выделенным дефицитарным ферментом. Инъекции выполняются пожизненно, позволяют восстановить метаболизм липидов.
- Субстратредуцирующая терапия. Лечение направлено на снижение интенсивности образования патологически накапливаемого соединения (сложного липида, его метаболитов). Используются молекулы с низкой массой, которые стимулируют остаточную активность фермента.
- Симптоматическая терапия. Препараты подбираются индивидуально на основании клинической картины болезни. Распространено применение антиконвульсантов, обезболивающих средств, ингибиторов АПФ, гепатопротекторов. При отдельных видах липоидозов использование симптоматических лекарств является единственным способом лечения.
Прогноз и профилактика
Липоидозы характеризуются гетерогенностью генетического дефекта и выраженной клинической полиморфностью. Большинство из них имеет непрерывное прогрессирующее течение. Правильная диагностика и своевременное лечение позволяют увеличить продолжительность жизни больных, а при легких формах способствуют улучшению адаптации. Профилактика возможна на этапе планирования и в первые месяцы беременности. Супружеским парам с высоким риском передачи болезни ребенку рекомендуется медико-генетическое консультирование, а в первом триместре – исследование амниотической жидкости и материала биопсии хориона на наличие мутаций генов.
1. Лизосомные болезни накопления липидов у детей/ Захарова И.Н. и др.// Медицинский совет. - 2016 - №1.
С момента описания в 1881 году первого такого заболевания, – а им стала ранняя детская амавротическая идиотия, или болезнь Тея-Сакса, – к настоящему времени выделено уже более полусотни лизосомных болезней накопления. При этом лизосомы как внутриклеточные органоиды были открыты гораздо позже, в 1955 г. (К. де Дюв, Бельгия), после чего механизм развития многих уже известных к тому моменту заболеваний значительно прояснился.
Заболеваемость находится в зависимости от этнических и территориальных факторов; каждая отдельная лизосомная болезнь накопления сама по себе является редкой (1:100 000 и реже), однако в совокупности эта группа заболеваний проявляется тем или иным своим вариантом у одного из 6-7 тысяч новорожденных.
Причины
Все классические ЛБН развиваются при наличии наследуемого дефектного гена, которым неправильно кодируется выработка какого-либо лизосомного энзима (фермента). Абсолютное большинство известных на сегодняшний день лизосомных болезней накопления, – за исключением трех, – наследуется по аутосомно-рецессивному типу. Это означает, что для появления признака носителями мутировавшего гена должны быть оба родителя; риск рождения больного ребенка в этом случае составляет 25%, вероятность пассивного носительства той же мутации ребенком – 50%, и еще 25% остается на шанс рождения ребенка здорового и не несущего каких-либо генетических сбоев. От пола эти вероятности не зависят.
Симптоматика
Когда речь идет о группе из нескольких десятков заболеваний, клиническая картина обычно оказывается чрезвычайно вариативной и полиморфной. В случае же с лизосомными болезнями накопления ситуация несколько иная: достаточно много общих черт. Так, все эти заболевания (мукополисахаридозы, сфинголипидозы, ганглиозидозы и т.д.) поражают ткани, в клетках которых присутствует большое количество лизосом – будь то паренхима селезенки, головной мозг, бронхолегочная система и пр. Типичными признаками выступают неуклонное прогрессирование симптоматики, гипертрофия органов (в особенности это касается печени и селезенки), грубые деформации костных элементов опорно-двигательного и челюстно-лицевого аппарата, а также, – в зависимости от разновидности ЛБН, – геморрагический, астенический, эпилептиформный, нефротический и другие синдромы. Тяжелейшие нарушения наблюдаются со стороны ЦНС; при манифестации в младенческом или раннем детском возрасте отмечаются глубокие задержки психофизического развития. Лизосомные болезни накопления приводят к ранней инвалидизации; в силу сложностей диагностики и лечения средняя продолжительность жизни пациентов с ЛБН гораздо ниже, чем в общей популяции.
Диагностика
Отсутствие патогномоничных клинических признаков и лабораторных коррелятов (в стандартных клинических и биохимических анализах), а также редкость заболеваний данной группы, – осложняют диагностику и повышают вероятность ошибочного диагноза. Важное значение имеет медико-генетический анамнез. Золотым стандартом сегодня являются критерии диагностики, базирующиеся на оценке активности ферментов (например, лизосомной кислой липазы). Стопроцентное подтверждение тому или иному типу ЛБН могут дать только методы генетического анализа.
Лечение
Помимо сугубо паллиативного, симптоматического лечения отдельных проявлений ЛБН, в последние годы обнадеживающие результаты связаны с заместительной ферментотерапией. Однако такие процедуры должны быть достаточно частыми и продолжаться пожизненно, что на сегодняшний день является весьма и весьма проблематичным. Есть основания полагать, что в обозримом будущем проблема лизосомных болезней накопления (и других наследственных заболеваний) будет решаться путем применения методов генной терапии и, возможно, генной профилактики. Это авангардное направление сегодня развивается быстро и, будем надеяться, приведет к очередной, крайне необходимой революции в медицине.
Читайте также: