
Лабораторная диагностика порфирий реферат
Обновлено: 05.07.2024
Рассмотрена классификация порфирий, описана клиническая картина заболевания, даны подходы к диагностике и сложности лечения (единственным патогенетическим методом лечения являются препараты гема). Приведено подробное описание клинического случая.
Complexities of diagnostics and treatment of porphyria polyneuropathy (selection of the clinical case)
Porfirius classification is examined, the clinical picture of disease is described, approaches to diagnostics and complexity of the treatment (only pathogenetic method of treatment they are the preparations of hem) are given. The detailed description of the clinical case is given.
Порфирии — это группа наследственных заболеваний, обусловленных нарушениями синтеза гема с повышением содержания промежуточных продуктов порфиринов в крови и тканях.
Гем является небелковой частью сложных белков гемпротеидов. По биохимическим функциям гемпротеиды делятся на неферментные (гемоглобин, находящийся в эритроцитах крови и клетках костного мозга, около 83%; миоглобин скелетных мышц и сердца, около 17%) и ферментные (цитохромы, каталаза, пероксидаза и др., менее 1%). Биологическая ценность гема состоит в том, что большинству белков, принимающих участие в обмене веществ, гем необходим в качестве простетической группы. Гем играет ключевую роль в связывании и транспортировке кислорода белками (гемоглобин, миоглобин). Гем, содержащийся в ферментах, принимает участие в процессах кумуляции энергии и окислительно-восстановительных процессах в клетках (цитохромы переносят электроны и входят в состав дыхательной цепи митохондрий и цепи микросом, цитохром Р-450 выполняет многочисленные функции, включая детоксикацию в печени; каталазы и пероксидазы участвуют в разложении пероксида водорода).
Характерной особенностью гемпротеидов является обмен небелковой части этих сложных белков. Биосинтез гема проходит восемь этапов; исходными веществами в синтезе служат глицин и сукцинил-КоА. В дальнейшем гем комплексируется с глобином с образованием гемоглобина или миоглобина. При синтезе других гемпротеидов гем соединяется с характерной белковой частью цитохромов и других гемсодержащих ферментов. Если белка недостаточно, чтобы связать образовавшийся гем, то происходит окисление гема с кислородом в гемин (комплекс протопорфирана IX с Fe 3+ ), который угнетает первую реакцию синтеза порфиринов.
Ферменты, участвующие в биосинтезе гема, имеются в костном мозге, ядросодержащих эритроцитах, печени, а также в почках и слизистой кишечника. В связи с чем порфирии подразделяются на эритропоэтические и печеночные в зависимости от ткани, где происходит преимущественное нарушение метаболизма порфиринов.
Классификация порфирий
1. Печеночные формы порфирий:
- порфирия, обусловленная дефицитом дегидратазы дельта-аминолевулиновой кислоты;
- острая перемежающаяся порфирия (ОПП);
- наследственная копропорфирия;
- вариегатная порфирия;
- поздняя кожная порфирия.
2. Эритропоэтические формы порфирий:
- врожденная эритропоэтическая порфирия;
- эритропоэтическая протопорфирия.
ОПП самая частая форма порфирий, распространенность в европейских странах составляет 7–12 случаев на 100 000 населения, и, как правило, она наиболее тяжело клинически протекает. У 80% носителей патологического гена ни разу в жизни не возникает каких-либо клинических проявлений (латентная, субклиническая порфирия). Лишь у 20% носителей патологического гена в течение жизни возникают клинически явные приступы ОПП.
Приступы могут спровоцировать: прием лекарственных веществ (в 70% случаев ОПП, перечень лекарств постоянно увеличивается), эндогенные колебания гормонального фона (менструация, беременность), инфекции, оперативные вмешательства, алкоголь, мышьяк, свинец, гипокалорийная низкоуглеводная диета.
Течение приступа ОПП вариабельно. Общая продолжительность приступа колеблется от нескольких дней до нескольких месяцев. Полиневропатия обычно развивается остро или подостро. В большинстве случаев симптоматика достигает максимального развития в течение 1–4 недель, но иногда фаза прогрессирования продолжается до 2–3 месяцев. Прогрессирование происходит непрерывно или ступенеобразно.
Клиническая картина
Первый приступ обычно развивается в возрасте от 15 до 35 лет (значительно реже у детей или лиц старше 50 лет). У женщин клинические проявления возникают примерно в 1,5–2 раза чаще, чем у мужчин. В типичных случаях приступ ОПП начинается с вегетативных симптомов, к ним присоединяются психические расстройства, а затем преимущественно моторная полиневропатия, но процесс может остановиться на любой из этих стадий.
Первым симптомом приступа почти во всех случаях бывает боль в животе. Отмечаются также боли в пояснице и конечностях, которые часто не укладываются в определенные зоны иннервации. Характер и локализация болей варьируют, но иногда бывают столь сильными, что это служит поводом для диагностики почечной колики, острого живота или кишечной непроходимости и проведения лапаротомии. Ошибочной диагностике острого живота может способствовать и часто наблюдающаяся тахикардия. Синусовая тахикардия (до 120–160 уд. в минуту) наблюдается в среднем в 60% случаев и почти во всех тяжелых случаях. У 15–20% больных отмечается субфебрилитет, однако выраженная гипертермия не характерна и, скорее, указывает на развитие интеркуррентной инфекции или абсцесса. Операция, особенно под барбитуровым наркозом, может приводить к быстрому развитию тяжелой полиневропатии.
В 60% случаев отмечается повышение артериального давления (АД) и иногда бывает весьма значительным, приводя к острой гипертонической энцефалопатии. Иногда возникают пароксизмальные нарушения сердечного ритма, которые могут сопровождаться коллаптоидными состояниями.
Те или иные психические расстройства (тревога, депрессия, гипоманиакальное состояние, нарушение сна и другие) развиваются во время приступа у подавляющего большинства больных ОПП. В тяжелых случаях развиваются делирий и угнетение сознания. Иногда психические расстройства бывают единственными или доминирующими проявлениями приступа ОПП. Зачастую на раннем этапе многим больным с порфирией ошибочно ставят диагноз истерии, маниакально-депрессивного психоза, шизофрении, деменции.
У 5% больных развиваются генерализованные судорожные припадки, которые обычно служат неблагоприятным прогностическим признаком. Их причиной, помимо предполагаемого нейротоксического действия предшественников порфиринов, могут быть острая гипертоническая энцефалопатия или гипонатриемия, обусловленная синдромом неадекватной секреции антидиуретического гормона (АДГ). Гипонатриемия возникает во время более трети приступов ОПП. В отдельных случаях наблюдаются мигренеподобные головные боли, преходящие нарушения зрения по типу гемианопсии или корковой слепоты.
Ярким, часто встречающимся маркером острых печеночных порфирий является выделение разных оттенков красной или бурой мочи (порфирины — органические пигменты, имеющие в кислых растворах ярко-пурпурный, а в щелочных — красно-коричневый цвет).
Но наиболее опасные осложнения порфирии связаны с полинейропатией, которая развивается в 10–60% приступов, часто через 2–4 дня после появления боли в животе или психических расстройств. Полинейропатия имеет преимущественно моторный характер — ее основным проявлением служит нарастающий вялый тетрапарез. Симптомы порфирийной полинейропатии отмечаются вариабельностью и динамичностью. В отличие от других аксональных полинейропатий, при порфирии первыми нередко вовлекаются не ноги, а руки (с развитием бибрахиального пареза), причем проксимальные отделы иногда страдают в большей степени, чем дистальные. В тяжелых случаях вовлекаются мышцы туловища, в том числе в 10% случаев — дыхательные мышцы. Поражение черепных нервов с развитием бульбарного синдрома, слабости мимических мышц, глазодвигательных нарушений также происходит лишь в тяжелых случаях и обычно на фоне выраженного вовлечения конечностей.
По мере прогрессирования полинейропатии симптомы раздражения вегетативной нервной системы сменяются симптомами выпадения: ортостатической гипотензией, фиксированным пульсом, ослаблением моторики желудочно-кишечного тракта, тенденцией к гипогидрозу (иногда с эпизодическим профузным потоотделением), затруднениями при мочеиспускании.
На высоте симптомов в 10–30% случаев, протекающих с тяжелой полинейропатией, наступает летальный исход. Он более вероятен, если заболевание не было вовремя распознано и назначались порфириногенные препараты. Непосредственными причинами летального исхода бывают внезапная смерть, часто связанная с нарушением иннервации сердца и гиперкатехоламинемией, паралич дыхательных мышц или тяжелый бульбарный синдром.
У выживших восстановление начинается спустя 2–3 недели после того, как полинейропатия достигает максимума. Полное восстановление наблюдается часто, но может растянуться на несколько лет, в течение которых у больных сохраняются парезы кистей и стоп, вегетативная дисфункция. На фоне восстановления могут происходить рецидивы, часто более тяжелые, чем первый приступ.
Все клинические проявления приступа ОПП объясняются вовлечением вегетативной нервной системы, дисфункцией периферической или центральной нервной системы. Однако механизмы поражения нервной системы остаются неясными. Определенное значение в патогенезе имеют сосудистые и нейроэндокринные нарушения. Аминолевулиновая кислота и порфобилиноген обладают непосредственным тоногенным влиянием на сосудистую стенку и гладкую мускулатуру; а локальный спазм сосудов может приводить к ишемии и сегментарной демиелинизации в периферической и центральной нервной системе. Во время обострения происходит повышение содержания в крови катехоламинов, иногда до уровня, наблюдаемого при феохромоцитоме. ОПП — одна из частых причин синдрома неадекватной секреции АДГ, который связан с поражением гипоталамуса и приводит к гипонатриемии и гипоосмолярности плазмы и, вследствие этого, к выраженным общемозговым проявлениям (угнетению или помрачнению сознания, эпилептическим припадкам). Поражение нервной системы и других органов и тканей также связано с цитотоксическим воздействием избытка порфиринов и их предшественников. С кровью порфирины разносятся по всему организму и попадают в кожу. Там они при инсоляции взаимодействуют с фотонами (фотохимические реакции), передают поглощенную энергию молекулам кислорода с образованием свободных радикалов (в частности, супероксидного радикала) и вызывают фототоксическую реакцию.
Для подтверждения диагноза определяют концентрацию порфиринов в моче и плазме, активность ферментов, участвующих в синтезе гема. Окончательным этапом диагностики больных, особенно асимптомных носителей порфирии и в фазе ремиссии, является проведение ДНК-анализа.
Приводим подробное описание клинического случая
Лечение больных порфирией представляет определенные сложности. Специфическую, патогенетическую, терапию провести невозможно ввиду отсутствия препаратов гема в России, а от введения 40% глюкозы до 1000 мл/сут отказывались врачи-реаниматологи из-за незнания подобной методики лечения и отсутствия опыта. Нейрометаболическая терапия проводилась крайне осторожно и была очень ограничена ввиду большого количества порфириногенных препаратов из числа лекарственных средств, применяемых в каждодневной практике врача. По данным ВОЗ список порфириногенных препаратов ежегодно пополняется. Препараты Актовегин® и Цераксон® одни из немногочисленных лекарственных средств, не входящих в список порфириногенных, и обладают высоким цитопротективным, мембраностабилизирующим и антигипоксантными свойствами по отношению к клеткам как центральной, так и периферической нервной системы. А как уже указывалось выше, все клинические проявления приступа ОПП объясняются дисфункцией периферической (полинейропатия) или центральной нервной системы (острая гипертоническая энцефалопатия). С учетом высокого риска развития у этих больных таких грозных осложнений, как пароксизмальные нарушения сердечного ритма и судорожные эпиприступы, особое значение имеет отсутствие подобных противопоказаний у препаратов Актовегин® и Цераксон®.
Общее состояние больного стабилизировалось, но подтвердить диагноз лабораторно не удавалось в связи с отсутствием методики исследования в г. Ростове-на-Дону. 22.10.00 г., несмотря на положительную неврологическую динамику на фоне неспецифической терапии (Актовегин®, Цераксон®) и энтерального приема глюкозы, отмечалось покраснение мочи. Выполнено исследование мочи (cito) — эритроциты не выявлены, что явилось косвенным подтверждением наличия в моче порфиринов или их промежуточных продуктов метаболизма, приведших к окраске мочи в красный цвет. 29.10.00 г., при непосредственном участии родственников больного, удалось организовать исследование мочи в Гематологическом научном центре РАМН: содержание общих порфиринов 0,31 мг/л (норма до 0,15 мг/л), порфобилиногенов 60,5 мг/л (норма 0–2,0 мг/л), дельта-аминолевулиновой кислоты 21,7 мг/л (норма 0,1–4,5 мг/л).
10.11.00 г. больной переведен в гематологическое отделение Областной клинической больницы № 1 с положительной динамикой — уменьшилась выраженность тетрапареза (мышечная сила в проксимальных отделах рук 2,5–3 балла, в кистях — 4 балла, в проксимальных отделах ног 3–3,5 балла, в стопах — 5 баллов, сухожильные рефлексы с верхних конечностей отсутствуют, появились коленные рефлексы d = s, ахилловы живые с двух сторон), регрессировали тазовые нарушения, дисфония, дыхание самостоятельное адекватное, без участия вспомогательной мускулатуры, уменьшилась дисфагия, вегетативная лабильность (АД 120/80 мм рт. ст., пульс 100 уд. в мин, синкопальных состояний не отмечалось, нормализовалась температура тела), больной самостоятельно передвигается в пределах палаты, сохраняется астенизация. После проведения лечения Нормосангом в дозе 3 мг/кг внутривенно капельно в течение трех дней состояние больного значительно улучшилось, к концу декабря 2010 г. неврологический дефицит практически полностью регрессировал.
Порфирии ‒ большая группа наследственных заболеваний, характеризующихся нарушением биосинтеза гема и накоплением его токсичных метаболитов. Клинические проявления крайне разнообразны – от светочувствительности и кожных высыпаний до болей в животе, полного паралича и острых психозов. Диагностика осуществляется с помощью молекулярно-генетических тестов, специальных лабораторных методов определения порфиринов и их предшественников в моче и кале, оценки активности ферментов в крови. Лечение заключается в мероприятиях, направленных на снижение образования токсических метаболитов, их выведение из крови, проведении симптоматической терапии и хирургических вмешательств.
МКБ-10
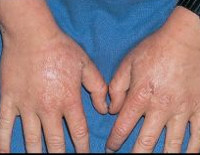
Общие сведения
Причины порфирий
В подавляющем большинстве случаев причиной порфирий выступают генетические мутации, обусловливающие неполноценность активности того или иного фермента, участвующего в биосинтезе гема. Исключением является поздняя кожная порфирия (спорадическая форма), которая развивается вследствие заболеваний печени (алкогольный гепатит, вирусный гепатит С) или длительной интоксикации тяжелыми металлами. Наследование порфирий происходит по аутосомно-доминантному или аутосомно-рецессивному типу. Синтезирование гема протекает в 8 последовательных этапов, за каждый отвечает свой фермент, кодируемый определенным геном. Для каждой формы порфирии существует специфичный ферментативный дефект.
Гем представляет собой комплексное соединение порфиринов с двухвалентным железом. Наибольшее количество гема образуется в печени и костном мозге. В печени гем входит в состав белков, участвующих в клеточном дыхании, расщеплении токсичных свободных радикалов и обезвреживании различных ксенобиотиков. В костном мозге гем используется для образования гемоглобина. Результатом сниженной активности ферментов является торможение синтеза гема на определенном уровне, что ведет к накоплению его токсичных промежуточных метаболитов.
Помимо генетической мутации, для развития острых порфирий необходимо воздействие провоцирующих факторов, стимулирующих выработку порфиринов. Такими факторами являются голодание, длительная инсоляция, стрессы, алкоголь, инфекции, интоксикация тяжелыми металлами (ртуть, свинец), лекарственные средства, подвергающиеся метаболизму системой цитохрома P-450 (нестероидные противовоспалительные препараты, антибиотики, антиконвульсанты, оральные контрацептивы, седативные средства). Особую роль играют колебания женских половых гормонов во время менструаций или беременности. У женщин месячные являются наиболее частым провоцирующим фактором, а беременность ассоциируется с тяжелым течением заболевания.
Патогенез
В результате неполноценности ферментов, участвующих в образовании гема, и действия провоцирующих факторов происходит увеличение концентрации его токсичных продуктов обмена. Для хронических порфирий характерно накопление протопорфирина, копропорфирина и упопорфирина. При острых формах возрастает количество порфобилиногена и дельта-аминолевулиновой кислоты (ДАЛК).
Порфирины накапливаются в коже и под действием ультрафиолетового излучения (солнечного света) запускают процесс перекисного окисления липидов, вызывая деструкцию и гибель клеток кожи. Копропорфирин и протопорфирин усиливают пигментацию кожи и ускоряют рост волос (гипертрихоз). Плохо растворимый в воде протопорфирин откладывается в клетках печени, закупоривает портальные тракты и желчные протоки. Отложение уропорфирина в эритроцитах приводит к их ускоренному разрушению в селезенке (гемолиз). Предшественники порфиринов (ДАЛК и порфобилиноген), накапливаясь в нервной ткани, вызывают демиелинизацию и аксональную дегенерацию нервных волокон.
Классификация
В основу разных классификаций порфирий положены различные критерии: клиническая симптоматика, локализация нарушения метаболизма порфиринов или тканевая тропность. Наиболее целесообразно выделять следующие виды порфирий:
- Эритропоэтические. К ним относятся врожденная эритропоэтическая порфирия (ВЭП, болезнь Гюнтера), эритропоэтическая протопорфирия (ЭПП). Основной клинический признак – поражение участков кожи, которые подвергаются воздействию прямых солнечных лучей (фотосенсибилизация). Данные патологии являются наиболее тяжелыми и имеют самый высокий процент летальности.
- Острые печеночные. Сюда включены острая перемежающаяся порфирия (ОПП), вариегатная порфирия (ВП), наследственная копропорфирия (НКП). Острые порфирии характеризуются приступообразным течением. Преимущественно страдает нервная система. При ВП и НКП также встречаются признаки фотосенсибилизации.
- Хронические печеночные. К ним относят позднюю кожную порфирию (ПКП), которая имеет наследственную и приобретенную формы. Это наиболее благоприятный вид порфирии.
Симптомы порфирий
Спектр клинических проявлений очень широк. Порфирии могут протекать в виде острых атак или хронически. Различия наблюдаются также в возрасте дебюта заболевания. Так, эритропоэтические порфирии манифестируют уже в дошкольном детстве (3-5 лет), острые порфирии - после полового созревания (14-16 лет), а спорадическая (приобретенная) форма ПКП - после 40 лет.
При острых порфириях развиваются сильные боли в животе, задержка стула, учащение сердцебиения, повышение артериального давления, изменение цвета мочи (от розового до красно-бурого). Тяжесть состояния пациента в основном обусловлена неврологическими симптомами – болью по всему телу, снижением чувствительности, прогрессирующей мышечной слабостью, иногда достигающей полного паралича, судорожными припадками, различными психическими расстройствами (тревожность, психомоторное возбуждение, бред, галлюцинации).
При поздней кожной форме возникает гиперпигментация участков кожи, подвергающихся постоянному воздействию солнечного света (лицо, шея, ушные раковины, верхняя часть груди, кисти рук). Кожа приобретает землистый или бронзовый оттенок. Также характерны гипертрихоз лобно-височной области лица, фотосенсибилизация, проявляющаяся повышенной ранимостью кожи и образованием пузырей с жидкостным содержимым. После вскрытия пузырей формируются эрозии. На местах разрешения эрозий образуются атрофические рубцы.
При эритропоэтических порфириях наблюдаются более выраженные признаки светочувствительности, чем при ПКП (ранимость, пузыри, эрозии). При длительном нахождении на свету появляется покраснение и сильное жжение кожи. Обширные эрозии оставляют после себя грубые рубцы на лице, что приводит к обезображиванию внешнего вида больного. В результате множественных рубцов на коже кистей рук развиваются контрактуры суставов, что значительно затрудняет их движения. Моча становится красной или розовой, а зубы окрашиваются в красно-коричневый цвет (эритродонтия). Из-за увеличенной селезенки могут появиться тяжесть или ноющие боли в левом подреберье. Специфический признак ЭПП – утолщение, огрубение и уплотнение кожи вокруг рта и глаз, на крыльях и спинке носа, на тыльных поверхностях кистей.
Осложнения
Нарушения порфиринового обмена ухудшают течение сердечно-сосудистых заболеваний, неблагоприятно влияют на углеводный метаболизм и повышают риск развития сахарного диабета 2 типа. Острые формы порфирий вследствие выраженной полинейропатии осложняются параличом дыхательной мускулатуры, аспирационной пневмонией, отеком головного мозга, тромбоэмболиями, рабдомиолизом. Постоянные эрозии кожных покровов могут привести к бактериальным инфекциям. При ЭПП из-за отложения нерастворимого в воде протопорфирина может развиться цирроз печени и печеночная недостаточность.
Диагностика
При подозрении на порфирию пациента направляют к врачу-гематологу. При постановке диагноза учитывается наличие заболевания у близких родственников, возраст больного, обстоятельства возникновения симптомов (инсоляция, прием лекарств или алкоголя, голодание, инфекции, менструации, беременность). Лабораторная диагностика порфирий следующая:
- Клинико-биохимические анализы. При болезни Гюнтера в общем и биохимическом анализах крови выявляются признаки гемолиза (пойкилоцитоз, анизоцитоз, сфероцитоз, ретикулоцитоз, повышение непрямого билирубина и сывороточного железа), увеличение печеночных трансаминаз. У больных с острыми порфириями отмечается снижение уровня глюкозы и натрия, при ПКП - увеличение сывороточного железа и ферритина. Также у 80% с ПКП выявляются положительные маркеры вируса гепатита С.
- Специфические исследования. Для диагностики острых порфирий широко используется скрининговая проба Эрлиха (при смешивании специального реактива с мочой она окрашивается в красный цвет). Для ОПП характерно повышение ДАЛК и порфобилиногена в моче, для ВП - протопорфирина в кале, для НКП – копропорфирина в кале и моче. При ПКП в моче увеличено содержание уропорфирина, в кале – копропорфирина. При ЭПП наблюдается высокая концентрация протопорфирина в эритроцитах и кале, при ВЭП - уропорфирина в моче, кале и эритроцитах. При люминесцентной микроскопии плазма дает красное флуоресцирование при ПКП и эритропоэтических порфириях.
Также для подтверждения диагноза проводится определение уровня ферментов цикла биосинтеза гема в эритроцитах, лимфоцитах или плазме - порфобилиногендезаминазы (ОПП), копропорфириноген-оксидазы (НКП), протопорфириноген-оксидазы (ВП), уропорфириногенсинтетазы (ВЭП), уропорфириногендекарбоксилазы (ПКП), феррохелатазы (ЭПП). Заключительным этапом диагностики является молекулярно-генетическое тестирование для выявления мутаций генов, кодирующих перечисленные выше ферменты. Данные исследования особенно эффективны для распознавания асимптомных форм порфирий.
Эритропоэтические порфирии дифференцируют с дерматологическими заболеваниями (буллезным пемфигоидом, вульгарной пузырчаткой), с гематологическими патологиями, протекающими со спленомегалией (лейкозами, лимфомами, аутоиммунными гемолитическими анемиями) с болезнями почек. ПКП дифференцируют с заболеваниями печени, гемохроматозом, надпочечниковой недостаточностью. Острые порфирии следует дифференцировать с хирургическими заболеваниями, сопровождающимися сильной болью в животе, неврологическими и психиатрическими патологиями.
Лечение порфирий
Пациентов с острыми и эритропоэтическими порфириями необходимо госпитализировать отделение гематологии. Лечение ПКП возможно как в стационаре, так и в амбулаторных условиях. На сегодняшний день не существует эффективных методов, полностью ликвидирующих нарушения обмена порфиринов. Основной упор делается на патогенетическую и симптоматическую терапию, а также на устранение провоцирующих факторов. Способы лечения зависят от вида порфирий:
- Острые. Для подавления образования порфобилиногена и ДАЛК применяют гем-аргинат, производные АТФ (аденил, рибоксин) и большие дозы глюкозы с дальнейшим переходом на высокоуглеводную диету. Для купирования вегетативной симптоматики используют октреотид, для ускорения восстановления миелина в нервных волокнах - витамины группы В. При менструалозависимых атаках эффективна овариосупрессивная терапия. С этой целью назначают агонисты гонадотропин-рилизинг гормона.
- Эритропоэтические. Данные порфирии очень плохо поддаются терапии. Основное лечение – это защита кожных покровов от солнечного света (окна со стеклом, не пропускающим ультрафиолет, закрытая одежда, фотозащитные кремы, прием бета-каротина). Необходимо обрабатывать эрозии антисептическими растворами для профилактики инфекционных осложнений. При выраженном гемолизе показана спленэктомия. В некоторых случаях болезни Гюнтера эффективной оказывается трансплантация костного мозга. При ЭПП дополнительно назначаются гепатопротекторы (урсодезоксихолевая кислота, адеметионин) и антицирротическая терапия.
- ПКП. С целью удаления порфиринов и избытка железа проводят плазмаферез и флеботомию (кровопускания). При противопоказаниях к данным процедурам назначаются аминохинолиновые (хлорохин) и комплексообразующие (дефероксамин) препараты. Для уменьшения всасывания порфиринов в желудочно-кишечном тракте применяют энтеросорбенты (активированный уголь). Также используются солнцезащитные кремы. При наличии гепатита С необходима противовирусная терапия интерфероном-альфа и рибавирином.
Прогноз и профилактика
В большинстве случаев порфирии являются тяжелыми заболеваниями с неблагоприятным прогнозом. При эритропоэтических формах продолжительность жизни составляет около 30 лет, смерть наступает от интеркуррентных инфекций. ЭПП часто приводит к циррозу печени. При атаках острых порфирий летальный исход наблюдается в 15-20%, основная причина смерти – паралич дыхательных мышц. При ПКП прогноз благоприятный, тяжелых осложнений не происходит. Для предупреждения рецидивов рекомендуется избегать провоцирующих факторов – инфекций, голодания, стрессов, длительной инсоляции, употребления алкоголя и определенных лекарственных средств. Людям, у которых в семье есть больной порфирией, необходимо определять активность ферментов цикла синтеза гема и проводить ДНК-диагностику для выявления генетических мутаций.
1. Заболевания внутренних органов при манифестных и латентных нарушениях порфиринового обмена/ Кривошеев Б.Н. и др. – 2014.
2. Диагностика и лечение острых порфирий: Клинические рекомендации национального гематологического сообществ/ под ред. Пустовойт Я.С., Кравченко С.К., Шмакова Р.Г., Савченко В.Г. - 2018.
3. Диагностическая роль отдельных синдромов и симптомов в семиотике острых порфирий/ Пустовойт Я.С., Галстян Г.М., Савченко В.Г.//Гематология и трансфузиология. – 2014 - №59(3).
НАЦИОНАЛЬНЫЕ КЛИНИЧЕСКИЕ РЕКОМЕНДАЦИИ
ДИАГНОСТИКА И ЛЕЧЕНИЕ ОСТРЫХ ПОРФИРИЙ
Москва 2018 г.
В дальнейшем были идентифицированы все восемь ферментов в этом цикле [5]. Установлено, что каждая нозологическая форма порфирий связана с дефектом активности одного из ферментов (кроме синтетазы дельта-аминолевулиновой кислоты (-АЛК)), закодированного в одном гене.

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
Классификация
Порфирии подразделяются на эритропоэтические и печѐночные в зависимости от ткани, где происходит преимущественное нарушение метаболизма порфиринов (см. классификация I) [10]. Вместе с тем, порфирии могут подразделяться на формы с поражением кожных покровов и острые, провоцируемые формы (см. классификация II).
- Поздняя кожная порфирия
Острые формы порфирий характеризуются яркой неврологической и вегетативно-сосудистой симптоматикой, в основе которой лежит полинейропатия. При порфирии, обусловленной дефицитом дегидратазы δ-аминолевулиновой кислоты и острой перемежающейся порфирии (ОПП) нет кожных проявлений. Это объясняется тем, что у них активными метаболитами являются предшественники порфиринов (δ-АЛК и порфобилиноген (ПБГ)), которые не имеют сродства к тканям дермы.
При снижении активности ферментов поздних этапов биосинтеза гема происходит накопление собственно порфиринов, избыток которых в дерме приводит к фототоксическим реакциям. Вследствие этого клиника кожных поражений характерна для таких острых порфирий как: наследственная копропорфирия (НКП) и вариегатная порфирия (ВП), а также поздней кожной порфирии (ПКП) и эритропоэтических порфирий.
Этиология и патогенез
Развитие различных форм порфирий связано с нарушениями цикла биосинтеза гема и имеет общие черты. В основе развития каждой формы порфирии лежит генетически обусловленное снижение или отсутствие активности определѐнного фермента в цепи биосинтеза гема (рисунок). Гены ферментов расположены на разных хромосомах и не имеют групповой сцепленности. Снижение активности фермента до 50% от нормы может не иметь клинических проявлений.
При ОП реализовать генетическое носительство и спровоцировать клиническую манифестацию заболевания могут:
-лекарственные препараты (НПВС, барбитураты, цефалоспорины сульфаниламиды и др. список которых представлен в приложении)
Перечисленные факторы приводят к повышенному потреблению конечного продукта цикла биосинтеза – гема (например, активация системы цитохрома Р-450) [10], либо оказывают непосредственное стимулирующее воздействие на активность первого фермента цикла биосинтеза – синтетазы -АЛК, что приводит к повышению еѐ активности (например действие прогестерона) [7], в результате чего ускоряется синтез всех промежуточных продуктов метаболизма порфиринов. На этапе участия дефектного фермента начинается избыточное накопление метаболитов в токсических концентрациях, что приводит к обострению заболевания. При ОП избыточное накопление -АЛК и ПБГ в тканях приводит к сегментарной демиелинизации нервных волокон с нарушением нервной проводимости. Токсическому воздействию подвержены все отделы нервной системы человека.
Периферическая сенсорно-моторная полинейропатия является следствием вторичной демиелинизации нервных волокон.
Вовлечение вегетативной нервной системы имеет следующий патогенез: - поражение абдоминальных вегетативных сплетений сопровождается спазмом сосудов брыжейки и нарушением моторики кишечника.
- ослабление активности n. vagus приводит к преимущественному влиянию на сердечно-сосудистую систему симпатического отдела; этому также способствует 10-ти кратное увеличением экскреции катехоламинов и нарушение функции барорецепторов артериальных сосудов.
Нарушение функции центральной нервной системы является следствием токсического воздействия предшественников порфиринов на нейроны головного мозга и развития длительного спазма артериол, гипонатриемии и гипергидратации, что приводит к тяжѐлым энцефалопатиям.
Поздняя кожная порфирия (ПКП) и эритропоэтические порфирии имеют хроническое течение с периодами обострений, которые могут вызвать:
Повышенная светочувствительность кожных покровов связана с фотохимическими реакциями, спровоцированными порфиринами. Избыток порфиринов в коже подвергается активному воздействию спектра солнечного излучения с длинами волн 400 – 410 нм, что приводит к образованию реактивных частиц, например, супероксид аниона, активирующего ксантин-оксидазу, и других метаболитов, повреждающих клетки базальной мембраны. Повторные атаки приводят к развитию нескольких слоѐв базальных мембран и образованию пласта кровеносных сосудов в поверхностных слоях дермы. Реактивные кислородсодержащие частицы также могут приводить к высвобождению гистамина из тучных клеток, усиливая явления фототоксичности.
Изменения кожи. Уропорфириноген – основной метаболит при поздней кожной порфирии стимулирует синтез фибробластами коллагена. При эритропоэтической протопорфирии жирорастворимый протопорфириноген откладывается в стенке сосудов дермы, приводя к их утолщению.
Пигментирование кожи и гипертрихоз наблюдаются в периорбитальных областях при поздней кожной порфирии и эритропоэтических порфириях, однако, механизм этих изменений до конца не изучен.
Эпидемиология
Порфирии не являются эндемичными заболеваниями и с одинаковой частотой встречаются среди населения всех континентов. Частота встречаемости острых форм порфирий (ОП) по различным оценкам составляет 7-12 случаев на 100 тысяч здоровых людей. В то же время частота бессимптомного носительства генетических дефектов, приводящих к ОП, составляет ~ 50-100 случаев на 100000 человек.
Клиническая картина
Cимптомы, течение
-эритема, волдыри на открытых участках кожи.
Первый приступ острых порфирий может развиться в возрасте старше 14-16 лет, значительно чаще у женщин. Начало заболевания острое, реже подострое. После воздействия порфириногенных факторов появляются боли в животе, конечностях, пояснице, тошнота, рвота. К концу второй недели заболевания появляется мышечная слабость, переходящая в парезы и параличи. Характерны тахикардия (до 110-130 артериальная гипертензия (до 180/100), выделение мочи с красноватым оттенком, неадекватное поведение и галлюцинации. При отсутствии лечения в течение 20-40 дней у больных могут развиться бульбарные нарушения и паралич дыхательной мускулатуры. В анализах крови нередко выявляется гипонатриемия.
Диагностика
Лабораторная диагностика острых порфирий, кроме порфирии обусловленной дефицитом дегидратазы --АЛК основывается на определении в моче избытка порфобилиногена с помощью:
- качественного скрининг-теста свежего образца мочи больного с использованием реактива Эрлиха по методу Watson-Schwartz. При наличии в моче избытка ПБГ образуется окрашенный продукт розово-красного цвета;
- количественного определения содержания ПБГ в моче (норма не превышает 2 мг/л).
При порфирии, обусловленной дефицитом дегидратазы-АЛК в моче, определяется высокая концентрация дельта-аминолевулиновой кислоты при нормальном содержании ПБГ.
Диагноз острой порфирии устанавливается на основании характерной клинической картины и высокого содержания ПБГ или -АЛК в моче [1,17].
Дифференциальный диагноз между ОПП и ВП или НКП основывается при измерении содержания общих порфиринов в кале [1,17]. В норме концентрация порфиринов в кале менее 200 нмоль/г сухого веса. Нормальная концентрация общих порфиринов в кале подтверждает диагноз ОПП. Повышение концентрации в несколько раз свидетельствует в пользу НКП или ВП. При исследовании плазмы спектрофлюориметрическим методом можно дифференцировать НКП от ВП.
Следующим этапом диагностики является определение активности ферментов в клетках крови. Это позволяет подтвердить диагноз у больных с клиническими проявлениями заболеваний и выявить бессимптомных носителей. Несмотря на распространѐнность и удобство диагностики бессимптомного носительства порфирий путѐм оценки активности специфического фермента в клетках, этот метод не является абсолютно достоверным [8,6].
Поэтому заключительным этапом диагностики порфирий у больных и, в особенности, у бессимптомных носителей является проведение ДНК- анализа [2].
Причиной поражения кожных покровов может быть повышенная светочувствительность, как следствие одной из форм порфирий. Тем не менее, следует провести дифференциальную диагностику кожной порфирии с другими дерматологическими заболеваниями.
- определение общих порфиринов в плазме и исследование спектра ее поглощения при флюоресцентной спектроскопии;
Для подтверждения диагноза ЭПП следует провести дополнительный тест - определение общих порфиринов в эритроцитах. При повышенной их концентрации следует измерить соотношение свободного и Zn-связанного протопорфиринов. В случае значительного превышения концентрации свободного протопорфирина подтверждается диагноз ЭПП.
Если результаты всех перечисленных тестов у пациента с активными кожными нарушениями будут нормальными, то диагноз порфирии будет маловероятным..
В случае же получения хотя бы одного или нескольких положительных результатов подтверждается диагноз кожной порфирии, форма которой определяется с помощью диагностических тестов.
Дифференциальный диагноз
- соматизация и синдром хронической усталости
Первичный этап диагностики, на практике, является самым сложным и ответственным для врача. Вариативность течения и симптоматики ОП (особенно при атипичном и моносимптомном вариантах развития) создают значительные трудности в своевременной постановке этого диагноза, нередко уводя врача в сторону ошибочных предположений. Выполнение же скрининг теста на наличие избытка ПБГ в моче всем обращающимся без анализа первичных жалоб нерационально с точки зрения временных и материальных затрат. Опытным путѐм, подтверждѐнным статистическими данными, выделены характерные для ранних сроков течения и специфичные для ОП симптомы. Разработана шкала, содержащая данные анамнеза и симптомы как характерные для клинического течения острых порфирий, так и наиболее часто ложно приписываемых ОП. Скрининг имеющихся у пациентов симптомов и сопоставление этих симптомов с симптоматикой, представленной в таблице (приложение 1)*, позволяет по сумме баллов судить о степени вероятности наличия порфирии у больного. Такой подход позволяет минимизировать число ятрогений, связанных с назначением пациентам с неустановленным диагнозом порфириногенных препаратов.
*В шкале представлены симптомы, встречающиеся при острых порфириях, которым присвоено определенное количество баллов, имеющих как положительные, так и отрицательные значения в зависимости от частоты их встречаемости при острой порфирии. Эти баллы суммируются, и если сумма баллов составляет менее 5 - диагноз острой порфирии маловероятен, если от 5 до 15 баллов – диагноз порфирии возможен, если более 15 баллов, то диагноз острой порфирии высоко вероятен.
Поражение кожных покровов при порфириях следует дифференцировать с прочими дерматологическими заболеваниями.
Лечение
1. Острые порфирии, цель лечения – предупреждение атак заболевания и развития необратимых изменений нервной системы.
Профилактика развития приступов ОП предусматривает ограничение воздействия на организм провоцирующих факторов, а именно:
- приѐма лекарственных препаратов с повышенной порфириногенной активностью, инсоляции развития бактериальных и вирусных инфекции (особенно HCV, HBV, CMV), состояния гипогликемии
- у женщин с доказанной связью между наступлением menses и частыми приступами – предупреждение менструаций.
Если менструальные циклы часто (три и более раз в год) провоцируют атаки ОП, репродуктивную функцию необходимо подавлять, для чего используют оральные контрацептивы – Ригевидон, Овидон; ГнРГ- Золадекс, синарел; андрогены - сустанон, андриол; овариоэктомия в отдельных случаях. Отмена овариосупрессии производится после достижения длительного бесприступного (несколько месяцев и более) течения заболевания.
Лечение начинают при наличии нескольких симптомов ОП (см. выше) и повышении показателей порфиринового обмена (по сравнению с предыдущими данными в динамике). При развитии острого приступа необходимо немедленное начало патогенетической терапии для подавления избыточного биосинтеза порфиринов:
2. Обеспечение избыточного поступления в орагнизм углеводов (200-600гр сухого вещ-ва глюкозы). 40%-1000мл в/в капельно в сутки, ежедневно, 2-4 недели [4].
3. Сандостатин в дозе 100-500 мкг/сут., подкожно, ежедневно на протяжении от 4-х недель до 6 месяцев в сочетании с плазмаферезами 6-10 сеансов [13].
4. Рибоксин 2%-10мл в разведении на 100-200 мл 0.9% раствора NaCl, в/в капельно 1-2 раза в сутки, ежедневно 2-4 недели.
К порфириям относится группа наследственных заболеваний, в основе которых лежат нарушения порфиринового обмена, ведущие к увеличению содержания в организме порфиринов или их предшественников. Порфирины синтезируются во всех клетках организма, преимущественно в костном мозге и печени, в которых они используются для образования гемоглобина и ферментов (цитохромов, каталазы, пероксидазы и других).
Патогенез (что происходит?) во время Порфирий:
Данные о распространенности различных форм порфирий отсутствуют. Имеются лишь сведения о вариегатной порфирии, встречающейся наиболее часто у лиц белой расы Южной Африки, потомков переселенцев из Голландии. Больные этой формой порфирий обнаружены в Финляндии.
Типы наследования и патогенетические особенности различных форм эритропоэтических и печеночных порфирий приведены при описании каждой из них в отдельности.
Среди эритропоэтических порфирий выделяют эритропоэтическую уропорфирию и эритропоэтическую протопорфирию. Среди печеночных - острую перемежающуюся порфирию, наследственную копропорфирию, вариегатную порфирию, урокопропорфирию.
Симптомы Порфирий:
Эритропоэтическая уропорфирия - редкое врожденное тяжелое заболевание, наследуется аутосомно по рецессивному типу, наблюдается в одном поколении иногда у нескольких детей, у родителей - гетерозиготных носителей патологического гена - клинические проявления болезни отсутствуют.
Заболевание может проявляться у новорожденных, появляется моча красного цвета, фотосенсибилизация (освобождающийся из эритроцитов уропорфириноген окисляется в уропорфирин и откладывается в коже), спустя несколько недель или месяцев после рождения на теле ребенка появляются пузыри с последующим развитием язв, которые на фоне антибиотикотерапии рубцуются, оставляя участки склерозированной кожи на лице и другой локализации. У ребенка нередко возникают контрактура суставов и слепота, отсутствуют волосы, ногти.
У больных обнаруживается увеличение селезенки. Выявляется гемолитическая анемия с внутриклеточным гемолизом, повышен уровень свободного билирубина сыворотки крови и содержание ретикулоцитов, расширен эритро-нормобластический росток в костном мозге. Укорочена продожительность жизни эритроцитов, часто снижена их осмотическая резистентность. Повышен уропорфирин и копропорфирин мочи и эритроцитов. Заболевание приводит к инвалидности и нередко к смерти в раннем детском возрасте.
Эритропоэтическая протопорфирия возникает в детстве, наследуется аутосомно-доминантно, в патогенезе - нарушение синтеза гема из протопорфирина, касающееся, по-видимому, части эритрокариоцитов, а также возможно увеличение синтеза а-аминолевулиновой кислоты.
У больных отмечается выраженное повышение чувствительности к солнечному облучению (отек кожи, зуд, покраснение, на местах ожогов возникают пузыри с последующим изъязвлением). В большинстве случаев заболевание протекает не так тяжело, как эритропоэтическая уропорфирия. Рубцы обычно не образуются. Содержание уропорфирина и копропорфирина в эритроцитах и в моче чаще нормальное, протопорфирина IX в эритроцитах - повышено. Уровень протопорфирина в плазме крови может быть также повышен. Ане-мизация вследствие гемолиза развивается редко, что обусловливается наличием двух популяций предшественников эритроцитов в костном мозге (неизмененной и содержащей значительно увеличенное количество протопорфирина). Иногда появляются геморрагии, связанные с отложениями гиалина в стенках сосудов и последующим их разрывом. Селезенка увеличивается редко.
Эритропоэтическая копропорфирия представляет собой крайне редкое заболевание, наследуется аутосомно-доминантно, напоминает по клинической картине эритропоэтическую протопорфирию. В эритроцитах содержание копропорфирина возрастает в 30-80 раз по сравнению с нормой, наблюдается экскреция больших количеств копропорфирина III с мочой и калом. Фотосенсибилизация выражена нерезко. Острый приступ болезни может быть спровоцирован приемом барбитуратов.
Острая перемежающаяся порфирия - одна из форм печеночных порфирий получила такое название в связи с тем, что хотя характерные тяжелые неврологические явления могут привести к смерти, но иногда они стихают и возникает ремиссия. Заболевание наследуется аутосомно-доминантно, патогенез его связан с нарушением активности фермента уропорфириноген-1-синтетазы и повышением активности синтетазы дельта-аминолеаулиновой кислоты, оказывающей токсическое влияние на нервную клетку.
Содержание порфиринов в эритроцитах нормальное. В моче обнаруживается повышенное количество уропорфирина I и II, а также копропорфирина III. В перирод обострения процесса в моче выявляется предшественник порфиринов - порфобилиноген. Уровень печеночной синтетазы, дельта-аминолевулиновой кислоты увеличен. Значительно повышен порфобилиноген мочи.
Наиболее частыми симптомами являются абдоминальные боли различной локализации, что нередко приводит к хирургическому вмешательству, наблюдаются тяжелые полиневриты, парестезии, психозы и коматозные состояния, повышение артериального давления и выделение розовой мочи. Летальный исход обычно обусловлен респираторными параличами, некоторые больные погибают в период комы или от кахексии. Обострение заболевания часто провоцируется беременностью, приемом лекарственных средств (барбитуратов, сульфаниламидов, анальгина). У родственников больных могут выявляться биохимические признаки болезни при отсутствии каких-либо клинических симптомов (латентная форма перемежающейся порфирии).
Наследственная копропорфирия наследуется по аутосомно-доминантному типу, часто протекает латентно. При этой болезни обнаруживается нарушение активности фермента копропорфириногеноксидазы, повышение синтеза дельта-аминолевулиновой кислоты в печени.
По клиническим проявлениям напоминает острую перемежающуюся порфирию. В моче в период обострения заболевания повышение количества дельта-аминолевулиновой кислоты и порфобилиногена не достигает такого высокого уровня как при острой перемежающейся порфирии. В моче и кале значительно увеличено количество копропорфирина.
Вариегатная порфирия наследуется аутосомно-доминантно. Патогенез, вероятно, связан с нарушением активности фермента протопорфириногена - оксидазы и повышением синтеза дельта-аминолевулиновой кислоты. Болезнь характеризуется признаками присущими острой перемежающейся порфирии. Иногда развивается почечная недостаточность. Боли в животе и неврологическая симптоматика возникают также при использовании определенных лекарственных препаратов - барбитуратов, сульфаниламидов, анальгина.
Урокопропорфирия (поздняя кожная порфирия) часто встречалась в России среди лиц, злоупотребляющих алкогольными напитками, перенесших гепатит, имеющих контакт с бензином и гепатотоксическими ядами. У большинства из них имеются нарушения функций печени. В моче увеличено в основном содержание уропорфирина, а содержание копропорфирина увеличено незначительно, обнаруживается фермент активности уропорфиноген карбоксилазы. Эти данные позволяют думать, что речь идет о приобретенной форме урокопропорфирии. В то же время у большинства родственников больных урокопропорфирией выявляется повышение содержания уропорфирина в моче и копропорфирина в кале, в нескольких семьях имеется по 2-3 больных урокопропорфирией. Отмеченные факты не дают оснований полностью исключить возможное наследование урокопропорфирии, по-видимому, по аутосомно-доминантному типу.
Урокопропорфирия характеризуется кожной симптоматикой (повышенной чувствительностью к солнечному облучению, механической травме, распространенным утолщением или истончением кожи, образованием пузырей преимущественно на тыльной поверхности кистей и лице с последующим развитием рубцов и др.). Морфологические изменения кожи выражаются в виде первичного поражения дермы, изменения в эпидермисе являются вторичными. Важным клиническим симптомом является увеличение печени, нередко с нарушением ее функционального состояния.
Диагностика Порфирий:
Острую перемежающуюся порфирию следует дифференцировать от свинцового отравления, сопровождающегося болями в животе, полиневритом, при котором в отличие от острой порфирии наблюдается гипохромная анемия с базофильной пунктацией эритроцитов, высокий уровень железа сыворотки крови; качественная проба на порфобилиноген чаще отрицательная.
Лечение Порфирий:
Современная клиника не располагает методами патогенетического лечения порфирии. Некоторый эффект при эритропоэтической уропорфирии наблюдается от спленэктомии, сопровождающейся удлинением продолжительности жизни эритроцитов и освобождением меньше уропорфирина в единицу времени разрушенных эритроцитов, что приводит к снижению фотосенсибилизации. Основной путь снижения клинических проявлений фотосенсибилизации больных при отдельных формах порфирии - защита от солнечного облучения.
При острой перемежающейся порфирии противопоказаны анальгин и транквилизаторы, вызывающие обострение болезни. При наличии болевого синдрома применяются наркотические средства, аминазин; при повышении артериального давления - индерал или обзидан; для уменьшения выработки порфиринов внутривенно вводят концентрированные растворы глюкозы (до 200 г/сут). Проводится лечение аденозин-монофосфатом (внутримышечно по 50-60 мг), рибоксином (по 200 мг внутрь, 3-4 раза в день).
К каким докторам следует обращаться если у Вас Порфирии:
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Порфирий, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору .

По словам доктора Дэвида Дольфина, известного специалиста по порфирии и другим болезням печени, люди, которых считали вампирами или оборотнями, могли страдать именно эти редким заболеванием.
Сегодня больных порфирией часто лечат путем инъекций гемоглобина. В Средние века уколы были невозможны, поэтому потребление больших объемов крови являлось единственным способом, которым человек мог получить дополнительный гемоглобин. Больные порфирией отчаянно желали достать кровь, поскольку из-за нехватки гемоглобина наступала смерть. Неудивительно, что среди таких больных распространены патологические изменения личности и слабоумие.
Каковы причины порфирии?
Симптомы заболевания
Суммируя опыт исследователей, занимающихся острыми порфириями, приводим наиболее характерные клинические симптомы этого заболевания:
боли в животе, как правило в эпигастральной или правой подвздошной областях;
приступообразный характер, иногда бывают постоянными;
продолжающимися несколько часов или дней;
запор, реже - понос;
стойкая синусовая тахикардия (до 160 ударов в минуту);
мышечная атония (затрагивает чаще мышцы конечностей и пояса);
боли в конечностях, голове, шее и грудной клетке;
потеря чувствительности (наиболее выражена в плечевой и бедренной областях);
поражение черепно-мозговых нервов;
нарушение тазовых функций;
двигательные расстройства в виде вялых парезов и параличей;
депрессивные и истерические компоненты;
спутанность сознания и дезориентация;
зрительные и слуховые галлюцинации;
Эритропоэтическая уропорфирия - редкое врожденное тяжелое заболевание, наследуется аутосомно по рецессивному типу, наблюдается в одном поколении иногда у нескольких детей, у родителей - гетерозиготных носителей патологического гена - клинические проявления болезни отсутствуют.Заболевание может проявляться у новорожденных, появляется моча красного цвета, фотосенсибилизация (освобождающийся из эритроцитов уропорфириноген окисляется в уропорфирин и откладывается в коже), спустя несколько недель или месяцев после рождения на теле ребенка появляются пузыри с последующим развитием язв, которые на фоне антибиотикотерапии рубцуются, оставляя участки склерозированной кожи на лице и другой локализации. У ребенка нередко возникают контрактура суставов и слепота, отсутствуют волосы, ногти.
У больных обнаруживается увеличение селезенки. Выявляется гемолитическая анемия с внутриклеточным гемолизом, повышен уровень свободного билирубина сыворотки крови и содержание ретикулоцитов, расширен эритро-нормобластический росток в костном мозге. Укорочена продожительность жизни эритроцитов, часто снижена их осмотическая резистентность. Повышен уропорфирин и копропорфирин мочи и эритроцитов. Заболевание приводит к инвалидности и нередко к смерти в раннем детском возрасте.
Эритропоэтическая протопорфирия возникает в детстве, наследуется аутосомно-доминантно, в патогенезе - нарушение синтеза гема из протопорфирина, касающееся, по-видимому, части эритрокариоцитов, а также возможно увеличение синтеза а-аминолевулиновой кислоты.
У больных отмечается выраженное повышение чувствительности к солнечному облучению (отек кожи, зуд, покраснение, на местах ожогов возникают пузыри с последующим изъязвлением). В большинстве случаев заболевание протекает не так тяжело, как эритропоэтическая уропорфирия. Рубцы обычно не образуются. Содержание уропорфирина и копропорфирина в эритроцитах и в моче чаще нормальное, протопорфирина IX в эритроцитах - повышено. Уровень протопорфирина в плазме крови может быть также повышен. Ане-мизация вследствие гемолиза развивается редко, что обусловливается наличием двух популяций предшественников эритроцитов в костном мозге (неизмененной и содержащей значительно увеличенное количество протопорфирина). Иногда появляются геморрагии, связанные с отложениями гиалина в стенках сосудов и последующим их разрывом. Селезенка увеличивается редко.
Эритропоэтическая копропорфирия представляет собой крайне редкое заболевание, наследуется аутосомно-доминантно, напоминает по клинической картине эритропоэтическую протопорфирию. В эритроцитах содержание копропорфирина возрастает в 30-80 раз по сравнению с нормой, наблюдается экскреция больших количеств копропорфирина III с мочой и калом. Фотосенсибилизация выражена нерезко. Острый приступ болезни может быть спровоцирован приемом барбитуратов.
Острая перемежающаяся порфирия - одна из форм печеночных порфирий получила такое название в связи с тем, что хотя характерные тяжелые неврологические явления могут привести к смерти, но иногда они стихают и возникает ремиссия. Заболевание наследуется аутосомно-доминантно, патогенез его связан с нарушением активности фермента уропорфириноген-1-синтетазы и повышением активности синтетазы дельта-аминолеаулиновой кислоты, оказывающей токсическое влияние на нервную клетку.
Содержание порфиринов в эритроцитах нормальное. В моче обнаруживается повышенное количество уропорфирина I и II, а также копропорфирина III. В перирод обострения процесса в моче выявляется предшественник порфиринов - порфобилиноген. Уровень печеночной синтетазы, дельта-аминолевулиновой кислоты увеличен. Значительно повышен порфобилиноген мочи.
Наиболее частыми симптомами являются абдоминальные боли различной локализации, что нередко приводит к хирургическому вмешательству, наблюдаются тяжелые полиневриты, парестезии, психозы и коматозные состояния, повышение артериального давления и выделение розовой мочи. Летальный исход обычно обусловлен респираторными параличами, некоторые больные погибают в период комы или от кахексии. Обострение заболевания часто провоцируется беременностью, приемом лекарственных средств (барбитуратов, сульфаниламидов, анальгина). У родственников больных могут выявляться биохимические признаки болезни при отсутствии каких-либо клинических симптомов (латентная форма перемежающейся порфирии).
Наследственная копропорфирия наследуется по аутосомно-доминантному типу, часто протекает латентно. При этой болезни обнаруживается нарушение активности фермента копропорфириногеноксидазы, повышение синтеза дельта-аминолевулиновой кислоты в печени.
По клиническим проявлениям напоминает острую перемежающуюся порфирию. В моче в период обострения заболевания повышение количества дельта-аминолевулиновой кислоты и порфобилиногена не достигает такого высокого уровня как при острой перемежающейся порфирии. В моче и кале значительно увеличено количество копропорфирина.
Вариегатная порфирия наследуется аутосомно-доминантно. Патогенез, вероятно, связан с нарушением активности фермента протопорфириногена - оксидазы и повышением синтеза дельта-аминолевулиновой кислоты. Болезнь характеризуется признаками присущими острой перемежающейся порфирии. Иногда развивается почечная недостаточность. Боли в животе и неврологическая симптоматика возникают также при использовании определенных лекарственных препаратов - барбитуратов, сульфаниламидов, анальгина.
Урокопропорфирия (поздняя кожная порфирия) часто встречалась в России среди лиц, злоупотребляющих алкогольными напитками, перенесших гепатит, имеющих контакт с бензином и гепатотоксическими ядами. У большинства из них имеются нарушения функций печени. В моче увеличено в основном содержание уропорфирина, а содержание копропорфирина увеличено незначительно, обнаруживается фермент активности уропорфиноген карбоксилазы. Эти данные позволяют думать, что речь идет о приобретенной форме урокопропорфирии. В то же время у большинства родственников больных урокопропорфирией выявляется повышение содержания уропорфирина в моче и копропорфирина в кале, в нескольких семьях имеется по 2-3 больных урокопропорфирией. Отмеченные факты не дают оснований полностью исключить возможное наследование урокопропорфирии, по-видимому, по аутосомно-доминантному типу.
Урокопропорфирия характеризуется кожной симптоматикой (повышенной чувствительностью к солнечному облучению, механической травме, распространенным утолщением или истончением кожи, образованием пузырей преимущественно на тыльной поверхности кистей и лице с последующим развитием рубцов и др.). Морфологические изменения кожи выражаются в виде первичного поражения дермы, изменения в эпидермисе являются вторичными. Важным клиническим симптомом является увеличение печени, нередко с нарушением ее функционального состояния.
ОСНОВНЫЕ КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ
Основные клинические проявления порфирии выражаются в периодически возникающих приступах нарушения функционирования нервной системы и повышения чувствительности кожи к солнечному свету. Кожа приобретает красно-коричневый оттенок, крайне истончается и лопается на солнце, покрываясь шрамами и язвами. При этом повреждаются хрящи ушей и носа, они сильно деформируются, что очень влияет на внешность человека.
Врожденная форма порфирии встречается довольно редко и характеризуется тяжелым течением с поражением кожи и гемолитической анемией. Болезнь проявляется у новорожденных красной мочой, позже образуются пузыри и язвы, которые очень плохо заживают, отягощаются вторичной инфекцией, со временем у ребенка зубы приобретают красноватое свечение, увеличивается селезенка.
У малыша с порфирией возникает контрактура суставов, слепота, отсутствуют волосы и ногти. Заболевание приводит к инвалидизации и смерти в раннем возрасте. Роль наследственного фактора в формировании болезни во взрослом возрасте отмечается как незначительная, чаще это влияние внешней среды, особенно для мужчин, старше 50 лет, склонных к употреблению алкоголя и длительное время пребывающих на солнце.
Острая перемежающаяся порфирия — это одна из форм печеночной порфирии, при которой наиболее распространенными симптомами являются интенсивные абдоминальные боли, тяжелые полиневриты, психозы, а также коматозные состояния и повышение АД. Прогноз заболевания зависит от степени повреждения печени, высока вероятность развития рака печени, цирроза, а также гемохроматоза. Возможный летальный исход обусловлен дыхательным параличом и остановкой сердечной деятельности.
Порфирия имеет весьма типичные внешние проявления:
Фотосенсибилизация или повышенная чувствительность к свету, проявляющаяся развитием дерматоза, напоминающим ожоговую болезнь. На бледной истонченной коже открытых участков тела появляются пигментация, пузыри, которые быстро вскрываются и обнажают эрозии и язвы, на месте которых образуются розово-синеватые рубцы, эрозии оставляют пигментацию.
Для порфирии характерен гипертрихоз и потемнение волос, шикарные бакенбарды, густые пушистые ресницы и брови.
Рубцовые изменения кожи в области ушей, носа, надбровных дуг, пальцев рук приводят к искажению черт лица и деформации кистей. На боковых поверхностях висков появляются микроцисты — последствия заживления мелких эрозий, напоминающие белые угри.
Под действием света изменяются, утолщаются, деформируются и разрушаются ногти, склерозируется ногтевое ложе.
У больных порфирией можно заметить типичные изменения глаз: красные склеры, конъюнктивит, помутнение роговицы, пигментация диска зрительного нерва и другие дистрофические процессы.
Порфирия нередко приводит к изменению десен и губ. Эрозивный хейлит проявляется покраснением, пигментацией, отечностью и кровоточивостью красной каймы губ. Атрофия слизистой десен приводит к обнажению шеек зубов и визуальному эффекту увеличения клыков.
Поверхность зубов и десен, на которые падает солнечный свет, приобретает красноватый оттенок, иногда становится темно-красной.
Поздними признаками порфирии является поражение сердечно-сосудистой системы, печени и других внутренних органов. Необратимые изменения часто приводят к смерти, летальность при острых атаках порфирии составляет примерно 20%.
ДИАГНОСТИКА.
Острую перемежающуюся порфирию следует дифференцировать от свинцового отравления, сопровождающегося болями в животе, полиневритом, при котором в отличие от острой порфирии наблюдается гипохромная анемия с базофильной пунктацией эритроцитов, высокий уровень железа сыворотки крови; качественная проба на порфобилиноген чаще отрицательная.
Сегодня медицина не располагает приемами и навыками лечения порфирии, замечено, что некоторое облегчение наблюдается при спленэктомии, переливании эритроцитарной массы, пересадке костного мозга, защите от солнечных лучей, воздержании от алкоголя, организации диеты питания, как для больных хроническим гепатитом.
Лечение Порфирий:
Современная клиника не располагает методами патогенетического лечения порфирии. Некоторый эффект при эритропоэтической уропорфирии наблюдается от спленэктомии, сопровождающейся удлинением продолжительности жизни эритроцитов и освобождением меньше уропорфирина в единицу времени разрушенных эритроцитов, что приводит к снижению фотосенсибилизации. Основной путь снижения клинических проявлений фотосенсибилизации больных при отдельных формах порфирии - защита от солнечного облучения.
При острой перемежающейся порфирии противопоказаны анальгин и транквилизаторы, вызывающие обострение болезни. При наличии болевого синдрома применяются наркотические средства, аминазин; при повышении артериального давления - индерал или обзидан; для уменьшения выработки порфиринов внутривенно вводят концентрированные растворы глюкозы (до 200 г/сут). Проводится лечение аденозин-монофосфатом (внутримышечно по 50-60 мг), рибоксином (по 200 мг внутрь, 3-4 раза в день).
ЖИЗНЬ С ПОРФИРИЕЙ.
История из жизни.
Журналист Владимир Лаговской пишет о случае порфирии у девочки, болеющей ею уже более 15 лет (фамилия и место жительства не называются). «Дневной свет смертелен для Kати. Солнце может сжечь ее заживо всего за одну минуту. Девочка больна. Но для окружающих не опасна. Kатина ночь длится уже 15 лет. С раннего детства на лице и ручках, открытых дневному свету, стали появляться пятна и сыпь. Девочка начинала кричать. Думали, что это обыкновенный диатез. Пройдет. Но пятна не проходили. Наоборот, превратились в язвы.
На луну девочка реагировала нормально. Не покрывалась язвами и при свете электрических лампочек. Для Kати убийственно лишь излучение определенной частоты - в основном ультрафиолетового спектра. Kремы от загара высокой степени защиты помогают ей, но ненадолго: минут на пять. Потом начинается нестерпимая боль. Раньше, когда девочка была маленькой, ее перевозили с места на место в закрытой сумке. Теперь Kате сшили специальный светонепроницаемый шлем с насосом, который подает воздух для дыхания. Даже летом девочка носит плотное пальто и перчатки. В школу она не ходит. Занимается с учителями дома в комнате со стеклами, не пропускающими ультрафиолет.
- Взрослые распространяют злобные слухи, - сетует Катина мама. - Будто бы моя дочь - вампир. И в любой момент может кого-нибудь покусать. Одна газета даже написала, что по ночам она воет. Радикального средства нет, а имеющиеся лекарства лишь облегчают заживление кожи.
Игорь Kурбатов, сотрудник гематологического центра РАМН, полагает, что Kатя и в самом деле потенциальный вампир.
- У девочки редкая так называемая порфириновая болезнь, - говорит он. - Ее причина была обнаружена лет десять назад. Но сам недуг существовал и раньше. В средневековых легендах о вампирах описаны люди, страдавшие именно порфириновой болезнью. Наши канадские коллеги под руководством профессора Дольфина считают, что ее вызывает дефект в генах. Мы до конца не уверены, но суть вот в чем. Человек насыщен пигментами, так называемыми порфиринами. Они входят, например, в состав крови - в гемоглобин, делающий ее красной. У больных естественная циркуляция порфиринов нарушается. Они скапливаются под кожей и под действием солнечного света приобретают разрушительную силу. Начинают выделать ядовитый атомарный кислород, который буквально разъедает окружающие ткани. Черты лица могут исказиться до неузнаваемости, сделаться пугающими. Люди прячутся от света и выходят на улицу только по ночам. Вполне возможно, что некоторые больные пили кровь. Но не с дьявольской целью, а как лекарство. Они интуитивно чувствовали, что нужно восполнить потерю гемоглобина. А прежде его можно было добыть только из человеческой крови, непосредственно ее употребляя. Это облегчало страдания. Но сегодня все это излишне, так как гемоглобин продается в аптеках.
ЗАКЛЮЧЕНИЕ.Порфирия - заболевание не заразное, а наследственное. Если оно есть хотя бы у одного из родителей, в 25 процентах случаев инфицированным окажется и ребенок.
Также среди причин возникновения недуга называют инцест, перенесенный гепатит С, а еще… злоупотребление алкоголем.
Есть сведения, что в Средние века страдающих порфирией пытались лечить свежей кровью, хотя пить ее совершенно бессмысленно, облегчения это не приносит.
Так как порфирия - пока неизлечимое заболевание, то говорить о ее лечении можно лишь весьма условно. Сегодня таким больным делают инъекции с препаратами крови.
- К сожалению, в нашей стране не уделяют этому вопросу должного внимания, - говорит Ярослав Пустовойт, старший научный сотрудник Гематологического научного центра РАМН, руководитель группы по изучению и лечению порфирии. - В отличие от Запада, у нас нет ни специалистов, ни мощных центров по данной проблеме.
Читайте также: