
Болезни с нетрадиционным типом наследования реферат
Обновлено: 30.06.2024
Некоторые родословные наследственных болезней не могут объясняться типичным менделирующим наследованием ядерных генов. Теперь известно, что они вызваны мутациями митохондриального генома и проявляют материнское наследование. Болезни, вызываемые мутациями в митДНК, демонстрируют множество необычных особенностей, происходящих из уникальных характеристик биологии и функции митохондрий.
Митохондриальный геном
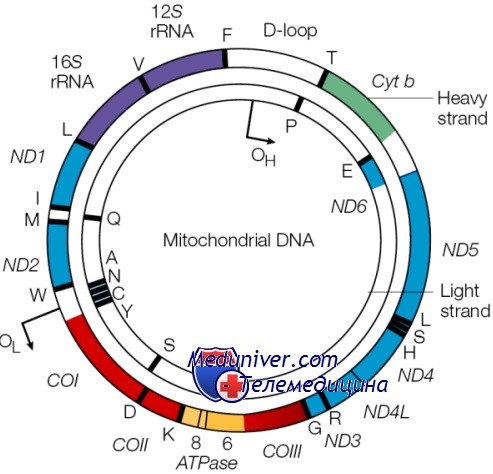
Не вся РНК и белок, синтезируемые в клетке, кодируются ДНК ядра; небольшая, но важная доля кодируется в генах митохондриального генома. Этот геном состоит из кольцевой хромосомы размером 16,5 килобазы, располагающейся в органеллах митохондриях, а не в ядре. Большинство клеток содержит по крайней мере 1000 молекул митДНК, распределенных по сотням отдельных митохондрий. Важное исключение — зрелый овоцит, имеющий более 100 000 копий митДНК, формирующих до одной трети общего содержания ДНК в этих клетках.
Митохондриальная хромосома содержит 37 генов. Они кодируют 13 полипептидов — компонентов ферментов окислительного фосфорилирования, два типа рРНК и 22 тРНК, необходимых для трансляции транскриптов генов митохондрий. Остальные полипептиды комплекса окислительного фосфорилирования кодируются ядерным геномом.
Репликативная сегрегация митохондриальной хромосомы
Первая уникальная характеристика митохондриальной хромосомы — отсутствие управляемой сегрегации, наблюдаемой в митозе и мейозе 46 ядерных хромосом. При делении клетки многочисленные копии митДНК в каждой митохондрии клетки копируются и произвольно расходятся во вновь синтезированные митохондрии. Митохондрии, в свою очередь, случайно распределяются между дочерними клетками. Этот процесс известен как репликативная сегрегация.
Гомоплазмия и гетероплазмия митохондриальной хромосомы
Вторая уникальная характеристика генетики митДНК возникает из-за того, что большинство клеток содержат много копий молекул митДНК. Когда мутация возникает в митДНК, она сначала присутствует только в одной из молекул в митохондрии. В ходе репликативной сегрегации митохондрия, содержащая мутантную митДНК, производит многочисленные копии мутантнои молекулы.
При делении клетка, содержащая смесь нормальных и мутантных митохондриальных ДНК, может передавать в дочерние клетки весьма различающиеся пропорции мутантнои и дикой митДНК. Одна дочерняя клетка может случайно получить митохондрии, содержащие чистую популяцию нормальных или чистую популяцию мутантных митохондриальных ДНК (ситуация, известная как гомоплазмия). Кроме того, дочерняя клетка может получить смесь митохондрий с мутацией и без нее (гетероплазмия).
Поскольку фенотипическая экспрессия мутации в митДНК зависит от относительных пропорций нормальной и мутантнои митДНК в клетках, формирующих различные ткани, неполная пенетрантность, переменная экспрессивность и плейотропия — типичные характеристики митохондриальных болезней.
Материнское наследование митохондриальной ДНК
Результат, определенный характеристиками генетики митДНК, называется материнским наследованием. Митохондрии сперматозоидов обычно отсутствуют в эмбрионе, поэтому митДНК наследуется от матери. Таким образом, все дети женщины, гомоплазмической по мутации митДНК, унаследуют мутацию, тогда как ни один из потомства мужчины, несущего ту же мутацию, не унаследует дефектную ДНК.
Материнское наследование гомоплазмической мутации митДНК, вызывающей наследственную нейропатию зрительного нерва Лебера.
Именно поэтому вариабельность процентного содержания мутантных молекул митДНК, обнаруживаемая в потомстве матери с гетероплазмией, возникает, по крайней мере частично, вследствие увеличения только части митохондриальных хромосом в овогенезе. Можно ожидать, что мать с высокой пропорцией мутантных молекул митДНК более вероятно произведет яйцеклетки с высокой пропорцией мутантных молекул митДНК, и, следовательно, более клинически пораженное потомство, чем мать с более низкой пропорцией. Есть одно исключение из материнского наследования, когда у матери имеется гетероплазмия по делеции в митДНК; по неизвестным причинам делеционная молекула митДНК обычно не передается от клинически больных матерей их детям.
Хотя митохондрии почти всегда наследуются исключительно через мать, существует, по крайней мере, один пример отцовского наследования митДНК у пациентов с митохондриальной миопатией. Следовательно, у пациентов с наблюдаемыми спорадическими мутациями митДНК должна учитываться редкая возможность отцовского наследования митДНК.
Точное определение семейной родословной — важная часть работы с каждым пациентом. Родословные могут демонстрировать как типичные менделирующие варианты наследования, так и более редкие, вызванные митохондриальными мутациями и половым мозаицизмом; или сложные варианты семейных случаев, не соответствующие ни одному из типов наследования. Определение типа наследования важно не только для установления диагноза у пробанда, это также идентифицирует других индивидуумов в семье, находящихся в группе риска и нуждающихся в обследовании и консультировании.
Несмотря на сложные цитогенетические и молекулярные анализы, используемые генетиками, точная семейная история, включая родословную семьи, остается фундаментальным средством для всех врачей и генетических консультантов, используемым при планировании индивидуального лечения пациентов.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Наследственные болезни человека это заболевания, связанные с нарушением работы наследственного аппарата клеток и передающиеся по наследству от родителей потомству. Основной резервуар генетической информации находится в ядерных хромосомах. Все клетки человеческого организма содержат в ядрах одинаковое количество хромосом. Исключение составляют половые клетки или гаметы — сперматозоиды и яйцеклетки, и малая часть клеток, которые делятся прямым делением. Меньшая доля генетической информации содержится в митохондриальной ДНК.
Наследственные болезни человека. Классификация.
Патология генетического аппарата бывает на хромосомном уровне, на уровне отдельного гена, а также бывает связана с дефектом или отсутствием нескольких генов. Наследственные болезни человека подразделяются на:
Хромосомные болезни
Наиболее известны хромосомные заболевания по типу трисомии — дополнительной третьей хромосомы в паре:
- Синдром Дауна — трисомия по 21 паре;
- Синдром Патау — трисомия по 13 паре;
- Синдром Эдвардса — трисомия по 18 паре хромосом.

Синдром Шерешевского — Тёрнера обусловлен отсутствием одной Х-хромосомы у женщин.
Синдром Кляйнфельтера — дополнительная Х-хромосома у мужчин.
Другие хромосомные болезни связаны со структурной перестройкой хромосом при их нормальном количестве. Например, потеря или удвоение части хромосомы, обмен участками хромосом из разных пар.

Генные болезни
Причины наследственных заболеваний на генном уровне заключаются в повреждении части ДНК, в результате которого возникает дефект одного определенного гена. Чаще всего генные мутации ответственны за наследственные дегенеративные заболевания или наследственные болезни обмена веществ в результате нарушения синтеза соответствующего структурного белка или белка-фермента:
- Муковисцидоз;
- Гемофилия;
- Фенилкетонурия;
- Альбинизм; ;
- Серповидноклеточная анемия;
- Непереносимость лактозы;
- Другие обменные заболевания.
Моногенные наследственные заболевания наследуются по классическим законам Грегора Менделя. Различают аутосомно-доминантный, аутосомно-рецессивный и сцепленный с полом типы наследования.

При близкородственных браках чаще всего реализуется именно генный тип наследственных заболеваний.
Заболевания с наследственной предрасположенностью или полигенные болезни
К ним относятся:
-
; ;
- Ишемическая болезнь сердца;
- Ревматоидный полиартрит;
- Рак молочной железы;
- Псориаз;
- Шизофрения;
- Аллергические заболевания;
- Язвенная болезнь желудка…
Список можно продолжать и дальше. Найдется лишь малая часть болезней, которые так или иначе не связаны с наследственной предрасположенностью. Действительно, все процессы функционирования организма обусловлены синтезом разнообразных белков, как строительных, так и белков-ферментов.

Но если при моногенных наследственных болезней за синтез соответствующего белка отвечает один ген, то при полигенных наследственных заболеваниях за сложный метаболический процесс отвечают несколько разных генов. Поэтому мутация одного из них может быть компенсированной и проявляться только при дополнительных внешних неблагоприятных условиях. Этим объясняется, что у больных данными заболеваниями дети болеют ими не всегда, и, наоборот, у здоровых родителей дети могут болеть этими болезнями. Поэтому в случае полигенных наследственных заболеваний можно говорить лишь о большей или меньшей предрасположенности.
Диагностика наследственных болезней
Методы диагностики наследственных болезней:

Однако, следует учитывать, что мутации генов BRCA1 и BRCA2 ответственны за рак молочной железы (РМЖ) только в 5-10%, а их наличие или отсутствие лишь изменяет степень риска достаточно редкой формы РМЖ. Расчет эффективности этого метода будет представлен в следующих публикациях.
Лечение наследственных болезней
Симптоматическое лечение заключается в коррекции метаболических и других патологических нарушений, связанных с данным заболеванием.
Диетотерапия направлена на исключение продуктов, содержащих вещества, которые не усваиваются или не переносятся больными.
Генотерапия направлена на введение в генетический аппарат клеток человека, эмбриона или зиготы генетического материала, компенсирующего дефекты мутированных генов. Успехи генотерапии пока невелики. Но медицина с оптимизмом смотрит на развитие генноинженерных методов в терапии наследственных заболеваний.
Чем опасны близкородственные браки?
Красивая, но недалекая блондинка обратилась к Эйнштейну: "Женитесь на мне, я рожу ребенка красивого, как я, и умного, как Вы". Эйнштейн благоразумно отказался. Действител.
Дальтонизм или цветовая слепота
Ночью все кошки серы! Действительно, при недостатке освещения цветовые рецепторы сетчатки глаза не работают, и мы видим лишь оттенки серого. Но стоит взойти солнцу, и кра.

В данной статье представлено описание и анализ наследования неменделевских наследственных болезней, значение и роль. Приведены примеры различных форм патологий (митохондриальные болезни, болезни импринтинга, экспансии тринуклеотидных повторов).
Ключевые слова: наследственные болезни, неменделевское наследование, наследование, болезни, митохондриальные болезни, болезни импринтинга, экспансии тринуклеотидных повторов.
В связи с развитием генетики известно достаточно большое число наследственных болезней, обусловленных изменением ДНК. Наследственные болезни в широком понятии — болезни, возникшие в результате различных дефектов и нарушений в наследственном аппарате клеток [1, c.126]. Мендель первый в биологии открыл фундаментальные законы наследственности. Простота и строгая модель этих законов, служила и служит опорой генетиков и является основой классической генетики. Однако, боле поздние исследования доказали, что далеко не все генетически контролируемые признаки подчиняются законам Менделя(на данный период времени считается, что не более половины всех случаев моногенной патологии четко соответствует законам Г. Менделя).
Неменделевская наследственность основана на явлениях эпигенетической изменчивости, при которой, в отличие от мутационной изменчивости, не изменяется генетический материал, и имеет наследуемые обратимые изменения генотипа. Неменделевское наследование возникает вследствие парамутаций, мейотического драйва, генной конверсии, экспансии микросателлитных повторов, инфицирования различными агентами (например, прионными белками) и других процессов. К таким феноменам относятся:
− Случайная инактивая одной из Х-хромосом в эмбриональном периоде развития — Х-половой хроматин.
− Хромосомные абберации, включающие унипарентную диполоидию, унипарентную дисомию и изодисомию.
− Экспансии тринуклеотидных повторов.
В менделевском наследовании каждый родитель вносит один из двух возможных аллелей для признака. Если генотипы обоих родителей в генетическом креста известны, законы Менделя могут быть использованы для определения распределения фенотипов, ожидаемых для населения потомства. Однако существует ряд явлений, которые изменяют фенотипы потомков так, что те не подчиняются классическим менделевским закономерностям:
− Разная вероятность образования гамет, их сочетания и жизнеспособность потомства.
− Однородительский тип наследования, при котором гены находятся не в ядре. Наследование генетического материала не зависит от вероятности расхождения хромосом в процессе мейоза. Главным образом это передача наследственной информации по материнской линии через цитоплазму яйцеклетки. Сперматозоид в зиготу передает только ядро с хромосомами. Следовательно, для признаков, гены которых находятся не в ядре,менделевские закономерности не работают. Вообще говоря, менделевские закономерности проявляются только у эукариот, размножающихся половым путем. Ни у прокариот, ни у бактерий они не работают.
− Взаимодействие аллелей между собой. Один аллель может подавлять другой (соотношение 1:3 во втором поколении) или не подавлять (тогда расщепление по фенотипу будет совпадать с расщеплением по генотипу 1:2:1).
− Взаимодействие генов между собой также может влиять на классическое менделевское расщепление. Один и тот же признак может контролироваться несколькими взаимодействующими неаллельными генами или разные гены могут приводить к одному и тому же фенотипу. Например, у глухих родителей может родиться слышащий ребенок, если гены, приводящие к такому фенотипу, у родителей были разные. Это типичная ситуация для всех моногенных болезней: если гомозиготность по какому-либо гену приводит к определенному признаку, то может быть другой ген, гомозиготность по которому приводит к таким же последствиям. Вероятность возникновения супружеской пары между людьми с гомозиготностью по одному и тому же гену очень мала. Поэтому рождение здорового ребенка от родителей, оба из которых имеют похожую аномалию, — нормальное явление. Кстати говоря, такие генетические меньшинства формируют субкультуры, и не редко родители хотят, чтобы их дети были такими же, как они. Кроме того, во многих культурах есть запрос на то, чтобы в потомстве появлялись только мальчики.
− Ограничение признака полом, например, наличие молочных желез у женщин и их отсутствие у мужчин. Хотя все гены для ее создания есть у обоих полов.
− Вероятность проявления аллеля при данном генотипе (пенетрантность). Проявление признака может быть незаметное или достаточно сильное.
− Влияние среды (не всегда формализуемое), например, окраска кролика, зависящая от температуры разведения [3].
В данной статье мы затронем только некоторые из болезней, вызванные следующими изменениями: митохондриальная, или цитоплазматическая, наследственность; болезни, связанные с экспансией тринуклеотидных повторов, и феномен геномного импринтинга.
В основном такой процесс проходит в 4 стадии:
1) ДНК-Полимераза сходится с прямым повтором и создает копию его.
2) ДНК-полимераза останавливает свою работу по тем или иным причинам, такие как отсутствие нужного нуклеотида.
3) Образуется отделение синтезированной цепочки, и на которой создается шпилька из повторов.
4) ДНК-Полимераза регенерирует свою работу, но из-за появившихся шпильки повторяет внесения дезоксирибонуклеотидов в позиции, к которым присоединила их до этого. Именно такой механизм находится в основе экспансии тринуклеотидных повторов [4].
Болезни экспансии подразделяются на две группы в зависимости от локализации тринуклеотидных повторов: в кодирующей части гена (болезнь Кеннеди, хорея Гентингтона) или некодирующей (атаксия Фридрейха, синдром Мартина-Белл). Особенности наследования таких заболеваний могут иметь как доминантный либо полудоминантный характер, проявления геномного импритинга и процесса антиципации, определяют сложности в их анализе и прогнозировании.
Хорея Гентингтона — хореическая деменция — аутосомный, доминантный тип, наследственное заболевание. Хорея появляется из-за повышения числа тринуклеотидных повторов (ЦАГ- цитозин, аденин, гуанин), которые отвечают за входящий в состав гентингтина глутамин. Цепочка аминокислот удлинняется, нарушая структуру гентингтина. Он соединяется (слипается) с другими белками, что приводит к гибели нейронов и нарушению работы ЦНС (центральной нервной системы).
Хорея — это гиперкинез, характеризующийся разбросанным беспорядочными подергиваниями мышц конечностей (особенно верхних), туловища и лица. Больные суетливы, гиперактивны, у них нарушена координация движений, они не могут усидеть на одном месте, для них типична танцующая походка. Хореические гиперкинезы возникают в возрасте 30–40 лет, позже к ним присоединяется быстро развивающееся слабоумие, доходящее до полного распада личности. Частая причина гибели суицид. [13]
Спинально бульбарная амиотрофия Кеннеди (болезнь Кеннеди) — это наследственное нейромышечное заболевание, которое со временем прогрессирует. Появляется эта болезнь из-за мутации андрогенового рецептора, находящийся на Х-хромосоме. Симптомы появляются после 40 лет, такие как уменьшение двигательных нейронов, дыхательная слабость, тремор, мышечные спазмы, потеря и атрофия мышц, эректильная дисфункция.
Атаксия Фридрейха — это наследственное заболевание, связанное с развитием дегенеративных процессов преимущественно проводящих систем спинного мозга (спинно — мозжечковых путей, задних столбов, пирамидных путей) и периферически нервных волокон. Характеризуется прогрессирующей атаксией, деформации скелета (позвоночника, грудной клетки, стоп и т. д.), дистрофическими изменениями миокарда. Наследуется по аутосомно-доминантному или аутосомно-рецессивному типу. Мутация происходит в гене FXN, кодирующем белок фратаксин. [13]
Проявляется болезнь постепенным нарушением походки: походка становится неуверенной, шаткой. Неустойчивость появляется в положении стоя, атаксия распространяется на мышцах рук и языка, речь- скандированная. Рефлексы отсутствуют или вызываются с трудом. Характерны деформации скелета — кифосколиоз и так называемая стопа Фридрейха. Наблюдается постепенное снижение психических функций. Кровь и церебральная жидкость без патологий. [13]
Синдром Мартина-Белл — генетическая патология с рецессивны типом наследования, связанный с ломкой Х-хромосомой т.е увеличение числа CGG-триплетных повторов (экспансия) в 5-нетранслируемой части гена FMR1 и, предположительно, дефицитом фолиевой кислоты. Болеют мальчики, среди умственно отсталых пациентов с этим синдромом около 6–10 %. Умственная отсталость варьирует от пограничной до степени имбецильности. У трети пациентов обнаруживается шизофреноподобные симптомы, такие как аутизм, вычурные и стереотипные движения, вращение вокруг оси тела, эхолалия, часто наблюдается синдром дефицита внимания, неврологическая патология, эпилептические припадки. [13]
Импринтинг (от англ. Imprinting — запечатление) — специфическая форма обучения, закрепление в памяти признаков объектов при формировании или коррекции врожденных поведенческих актов.
Геномный импринтинг — зависимость характера экспрессии генов у потомства, опирающаяся на то, от кого из родителей получены гены. Происходит независимо от законов Менделя.
Впервые различия между хромосомами, полученными от отца или от матери, были обнаружены учеными, работавшими в Филадельфии и Кембридже, в 1984 году.
Механизмы: метилирование нуклеотида с цитозином (ЦпГ-метилирование); метилирование аденина (ЦпА-метилирование).
Отсутствие метилированного цитозина сопутствует активации центра импринтинга, что приводит к различным заболеваниям (синдром Прадера-Вилли, синдромы Беквита-Видемана и Рассела-Сильвера).
У человека импринтинг проявляется в виде полного пузырного заноса. Если к зиготе перешли только мужские хромосомы, то развивается избыточный трофобласт, а эмбрион отсутствует или погибает. При наследовании только материнских хромосом происходит недостаточное развитие ворсин хориона, приводящее к гибели зародыша.
На данный момент о импринтированных генах известно, что:
- Большое количество импринтированных генов не являются ЗРК — кодирующими. Для многих из них механизм функционирования неизвестен.
- Огромное количество онкогенов и генов подавляющих развитие опухолей являются импринтированным. Передача генов является возможной от обоих родителей.
- Импринтированные генов влияют на метаболизм, работу щитовидной железы, выработку инсулина, а также непосредственно влияют на скорость роста плода и плаценты.
Аномалия геномного импринтинга — пузырный занос (дегенерация ворсин хориона). Различают полный (возникает при оплодотворении безъядерной яйцеклетки двумя спермиями; возможно образование опухолей — хорионэпителиом) и частичный пузырный занос (образуется в результате оплодотворения яйцеклетки двумя сперматозоидами). При частичном пузырном заносе отсутствует риск развития хорионэпителиомы.
Для хромосомного уровня геномного импринтинга характерны однородительские дисомии (ОРД). Различают гетеродисомию (в диплоидном наборе две гомологичные хромосомы одного родителя, с различными последовательностями ДНК) и изодисомию (в диплоидном наборе две гомологичные хромосомы одного родителя, с различными последовательностями ДНК). [11]
Три основные механизма формирования ОРД:
- Редукция одной из 3-х гомологичных хромосом в трисомной зиготе.
- Дупликация одной хромосомы, полученной от родителя.
- Слияние односомной гаметы с дисомной.
Геномный импринтинг является причиной следующих наследственных заболеваний человека:
Синдром Сильвера-Рассела. Причина развития — однородительская дисомия седьмой хромосомы. Болезнь начинает развиваться внутриутробно, на 6–7 неделях беременности. Наблюдается задержка роста, отставание в массе, недоразвитость наружных половых органов, ассиметрия головы, маленькое треугольное лицо с большим лбом и увеличение мозгового черепа. Частота заболевания составляет 1 на 20000 новорожденных.
Синдром Беквита-Видемана. Причина развития — дисомия 11-ой отцовской хромосомы. Признаки: большой рост, макроглоссия, омфалоцеле и гигантизм. После восьми лет рост замедляется. Характерна асимметрия тела, которая также распространяется и на внутренние органы — спланхномегалия, умеренная микроцефалия или гидроцефалия. Велика вероятность образования опухолей. Частота заболевания составляет 1 на 15000.
Синдром Прадера-Вилли. Причина развития данного — микроделеция 15-ой материнской хромосомы. Для заболевания характерны: избыточный вес, отставание в росте, гипогонадизм, умственная отсталость, нарушение координации, затрудненная речь, уровень интеллекта ниже среднего, внутриутробно наблюдается слабое шевеление плода. К году формируется гиперфагия, продолжительность жизни 25–30 лет. Частота заболевания составляет 1 на 20000. [10]
Помимо экспансии выделяют митохондриальные болезни. Митохондрии — электростанция в наших клетках, это значит, что главная функция — выработка энергии в форме АТФ. Впервые митохондриальные заболевания описал Люфт в 1962 году, когда обследовали 35-летнюю женщину с чрезмерным потоотделением, слишком высокой температурой. Её беспокоили разобщения процессов окислительного фосфорилирования. ОФ-основной источник получения энергии в клетке. Это привело к образованию тепла без образования АТФ, что влечет за собой нарушения функций состояния пациентки. В целях компенсации ее митохондрии увеличились в размере и умножились, что отчетливо наблюдалось при анализе гистологической мышечной биопсии. [1]
После этого случая было отмечено, что митохондриальная дисфункция приводит к патологическим состояниям. К таким состояния относится болезнь Паркинсона, болезнь Альцгеймера. [13]
Таким образом, рассмотрев все вышеперечисленные заболевания и механизмы их развития, можно с точностью утверждать, что они относятся к немеделевскому типу наследования, а геном человека — удивительная вещь, которую не объяснят только одни законы Менделеева.
Основные термины (генерируются автоматически): болезнь, ген, геномный импринтинг, повтор, причина развития, частота заболевания, родитель, хромосома, генетический материал, диплоидный набор.
Ключевые слова
болезни, наследование, наследственные болезни, неменделевское наследование, митохондриальные болезни, болезни импринтинга, экспансии тринуклеотидных повторов
наследственные болезни, неменделевское наследование, наследование, болезни, митохондриальные болезни, болезни импринтинга, экспансии тринуклеотидных повторов
Похожие статьи
Механизм формирования филадельфийской хромосомы и ее.
Филадельфийская хромосома (Ph-chromosome) является наиболее частой цитогенетической аномалией, обнаруживающейся в зрелых В-клетках пациентов с хроническим миелоидным лейкозом (ХМЛ), причем встречаемость этой мутации растет с возрастом.
Полиморфизм генов гемостаза у мужчин и женщин с бесплодием
Определены частоты частоты аллелей и генотипов в трех генах фолатного обмена (MTHFR
Большое влияние на нормальное развитие репродуктивной функции мужчин и женщин
Материалом для исследования служила венозная кровь, забранная Vacuette с ЭДТА из.
Генетика человека. Что определяют наши гены | Статья в журнале.
Однако бурное развитие генетики сделала ее труднопонимаемой для неспециалистов.
Возможность лечения генетических болезней. Генетика человека— это наука, объединяющая в себе генетику и медицины
Ген— это структурно-функциональная единица наследственности.
DA-система как генетический маркер заболеваний, связанных.
Библиографическое описание: Румянцева, В. Д. DA-система как генетический маркер заболеваний
Ген DRD2 расположен на плече хромосомы 11 в области q22.3–23.1, имеет длину около
Причиной этого является недостаточное количество положительных эмоций, а.
Изучение гетерогенности гена NPHS2 у детей со. | Молодой ученый
Наличие мутаций в гене NPHS2, кодирующий данный белок, приводит к развитию стероидрезистентного нефротического синдрома. Нами была исследована нуклеотидная последовательность гена NPHS2 в российской популяции детей.
Генетические аспекты болезни Паркинсона | Статья в журнале.
Актуальность. Болезнь Паркинсона (БП) — второе по частоте (после болезни Альцгеймера) нейродегенеративное заболевание, встречающееся практически повсеместно. Заболевание развивается чаще всего у лиц страше 60 лет, причем чаще страдают мужчины.
Генетические аспекты рассеянного склероза | Статья в журнале.
Рассеянный склероз является классическим мультифакториальным заболеванием, в этиологии которого важную роль играет взаимодействие внешних факторов и особенностей структуры большого количества генов. Выявление факторов риска развития рассеянного склероза и.
Значение гиперандрогенемии в причинах бесплодия
Актуальность данной темы базируется на частоте развития бесплодия на фоне гиперандрогении. Среди женщин с жалобами на гипоменструацию, невынашивание и наконец, бесплодие около 30 % страдают высоким уровнем андрогенов в крови.
Изучение популяционной структуры человечества для выявления.
Ключевые слова: наследственные заболевания, популяционная структура человечества
Болезнь Тея-Сакса, Тея-Гоше, контролируемые рецессивными генами, встречаются
С развитием средств массового перемещения людей на планете остается всё меньше.
Изменения в представлении о патогенезе Ph-негативных.
Безусловно, открытие выше указанных генетических нарушений при ХМПЗ, значительно улучшило наше понимание их развития, но осталось еще много неясного. Современные представления о патогенезе редко встречающихся ХМПЗ.
Генетические заболевания – это большая группа болезней человека, вызванных патологическими изменениями в генетическом аппарате. В настоящее время известно более 6 тысяч синдромов с наследственным механизмом передачи.
Виды генетических заболеваний человека

Основу наследственных заболеваний составляют генные, хромосомные и митохондриальные мутации.
Аутосомно-рецессивный. В этом случае происходит полная замена здоровых генов на мутантные. Ребёнок должен получить по одной копии рецессивного мутантного гена от каждого из родителей. У отца и матери может не наблюдаться данного заболевания, но это не исключает их как носителей гетерозиготной мутации. Вероятность, что у пары появится ребёнок с аутосомным рецессивным заболеванием равна 25%. Примеры: альбинизм, муковисцидоз.
Кодоминантный. Этот тип наследования подразумевает проявление и доминантного, и рецессивного гена, поэтому заболевание наследуется частично. Яркий пример: серповидно-клеточная анемия.
Наследование, сцепленное с полом. Означает, что наследование признаков передаётся только определенному полу. Например, гемофилией болеют исключительно мужчины.
Хромосомные болезни
Патологические изменения могут возникать как при потере генетического материала (например, при выпадении целой хромосомы или её части), так и при добавлении новых хромосом. Клинически характеризуется множественными врождёнными пороками развития. В настоящее время известно более 1000 хромосомных аномалий.
Точные причины возникновения до конца не изучены. Учёные предполагают, что провоцирующими факторами можно назвать ионизирующее излучение, химические вещества, вирусы, приём некоторых лекарств во время беременности, курение, алкоголь, возраст матери.
Хромосомные болезни могут быть связаны с нарушением:
1) числа хромосом;
3) структуры хромосом.
Общей чертой для хромосомных заболеваний является многофакторность поражения. А именно: пороки внутренних и наружных органов, черепно-лицевые дизморфии, замедленный рост и развитие, психическое и умственное отставание от сверстников, нарушение работы многих систем организма.
Перечислим некоторые из заболеваний:
Синдром кошачьего крика (делеция в 5-ой хромосоме);
Синдром Дауна (трисомия по 21-ой хромосоме);
Синдром Патау (трисомия в 13-ой хромосоме);
Генные мутации

Генные (точечные) мутации – это те, что возникают в результате изменения химической структуры гена и представляют собой замену, удаление или вставку нуклеотида. Возникают чаще, чем хромосомные и геномные, однако в меньшей степени меняют структуру ДНК. Также к генным мутациям относятся транслокации (перенос), дупликации (повторение), инверсии (переворот на 180°) участков гена, но не хромосомы.
рассмотрим мутацию ГТТ ЦЦЦ ГГТ → ГТЦ ЦЦЦ ГГТ.
В первом триплете произошла тимина заменился на цитозин. Триплеты ГТТ и ГТЦ кодируют глутаминовую кислоту, поэтому данная мутация не вызвала изменений в структуре белка: глу-гли-про → глу-гли-про.
В других же случаях замена нуклеотида может изменить порядок аминокислот в молекуле белка и привести к фенотипическим последствиям.
ГТТ ЦЦЦ ГГТ → ГТГ ЦЦЦ ГГТ.
В первом триплете тимин заменился на гуанин. ГТТ кодирует глутаминовую кислоту, а ГТГ — гистидин. Соответственно, первичная структура белка изменяется: глу-гли-про → гис-гли-про. Существует большая вероятность появления фенотипических изменений.
Мультифакториальные генетические болезни
Мультифакториальными генетическими заболеваниями называют патологии, возникающие при сочетании генетической предрасположенности и влиянии окружающей среды. Простой пример: пациент предрасположен к раку лёгких + в течении нескольких лет злоупотребляет курением. Соответственно, риск возникновения заболевания увеличивается в 2 и более раз.
К наиболее часто встречающимся мультифакториальным болезням относятся псориаз, цирроз печени, ревматоидный артрит, ишемическая болезнь сердца, бронхиальная астма.
Диагностика наследственных болезней

Жизнь человека начинается с момента зачатия. Чтобы уточнить состояние плода, важно провести пренатальную диагностику во втором триместре беременности. Тест поможет рассчитать риски различных синдромов (Дауна, Эдвардса, Корнели де Ланге) и дефектов.
Для определения метаболитов, специфических для наследственных болезней нарушения обмена веществ (энзимопатий), проводятся специальные пробы:
- проба на гипераминоацидурию;
- микробиологический тест Гатри.
Чтобы диагностировать наследственные нарушения обмена аминокислот, олигосахаридов и гликозамимногликанов (мукополисахаридов), используются более сложные методы аналитической биохимии;
- газовая и жидкостная хроматография;
- магнитная резонансная спектроскопия.
Помимо этого, медицина предрасполагает и другими методами определения генетических заболеваний:
Читайте также: