
Болезнь и синдром брутона реферат
Обновлено: 05.07.2024
Оглавление
Введение 3
Иммунодефицитные состояния 4
Первичные иммунодефициты 7
Вторичные (приобретенные) иммунодефициты. 9
Болезнь Брутона 14
Причины 14
Патофизиология 14
Симптомы и проявления 15
Диагностика 18
Лечение 19
Осложнения 21
Профилактика 21
Заключение 22
Список литературы 23
Введение
Иммунодефицитные состояния (ИДС) –стойкие или временные изменения иммунного статуса, обусловленные дефектом одного или нескольких механизмов иммунного ответа на антигенное воздействие.
В основе развития иммунодефицитных состояний и иммунодефицитов, как правило, находятся отсутствие или дефицит клеток иммунной системы и/или расстройства их функций. Это обусловливает высокую частоту развития при иммунодефицитах различныхинфекционных, паразитарных, опухолевых и аллергических заболеваний. С другой стороны, при истощающих заболеваниях часто развиваются иммунодефицитные состояния.
ЭТИОЛОГИЯ
Первичные иммунодефициты (наследуемые и врождённые дефекты иммунной системы) проявляются развитием инфекционных поражений организма вскоре после рождения, но могут не иметь клинических проявлений и до более позднего возраста.
Причина:
Генные и хромосомные дефекты (многочисленные иммунодефицитыразных классов).
Вторичные иммунодефициты, или иммунодефицитные состояния - иммунная недостаточность развивается вследствие эндо- и экзогенных воздействий на нормальную иммунную систему (например, около 90% всех вирусных инфекций сопровождается транзиторной иммунодепрессией).
Причины иммунодефицитных состояний многообразны, к ним отнесены:
-Иммуносупрессивные препараты (включая фенитоин, пеницилламин,глюкокортикоиды).
- Недостаточность питания, полостного и мембранного пищеварения, а также кишечного всасывания.
- Наркотики и токсические вещества.
- Лучевые воздействия, химиопрепараты.
-Рост злокачественных опухолей.
- Вирусы (например, ВИЧ).
- Состояния, приводящие к потере белка (например, нефротический синдром).
- Гипоксия.
- Гипотиреоз.
- Уремия.
- Аспления.
Для цитирования: Касохов Т.Б., Цораева З.А., Мазур А.И. и др. Первичный иммунодефицит гуморального звена иммунитета – болезнь Брутона у шестилетнего ребенка (клинический случай) // Эффективная фармакотерапия. 2019. Т. 15. № 40. С. 16–19.
- Аннотация
- Статья
- Ссылки
- Английский вариант
Первичный иммунодефицит представляет значимую проблему здравоохранения. Этим, в частности, обусловлено создание по инициативе ряда стран Европейского регистра пациентов с первичными наследственными иммунодефицитными состояниями и генных мутаций как причин первичной иммунной недостаточности. В детском возрасте заболевание чаще встречается у мальчиков, во взрослом – выявляется с одинаковой частотой у лиц обоего пола.
В статье рассмотрен клинический случай в отношении шестилетнего мальчика с болезнью Брутона. Болезнь Брутона (агаммаглобулинемия) – очень редкое (соотношение новорожденных мальчиков 1:1000000) Х-сцепленное наследственное заболевание, которое ассоциируется с мутацией гена цитоплазматической тирозинкиназы (Вtk), обеспечивающей дифференцировку пре-В-лимфоцитов. Данная патология характеризуется агрессивным течением. В отсутствие адекватной терапии возможен летальный исход.
- КЛЮЧЕВЫЕ СЛОВА: болезнь Брутона, иммунитет, иммунодефицит, иммуноглобулины, гуморальное звено
Первичный иммунодефицит представляет значимую проблему здравоохранения. Этим, в частности, обусловлено создание по инициативе ряда стран Европейского регистра пациентов с первичными наследственными иммунодефицитными состояниями и генных мутаций как причин первичной иммунной недостаточности. В детском возрасте заболевание чаще встречается у мальчиков, во взрослом – выявляется с одинаковой частотой у лиц обоего пола.
В статье рассмотрен клинический случай в отношении шестилетнего мальчика с болезнью Брутона. Болезнь Брутона (агаммаглобулинемия) – очень редкое (соотношение новорожденных мальчиков 1:1000000) Х-сцепленное наследственное заболевание, которое ассоциируется с мутацией гена цитоплазматической тирозинкиназы (Вtk), обеспечивающей дифференцировку пре-В-лимфоцитов. Данная патология характеризуется агрессивным течением. В отсутствие адекватной терапии возможен летальный исход.
Первичные иммунодефицитные состояния, или первичные иммунодефициты, как правило, наследуемые дефекты. Однако в настоящее время встречаются врожденные иммунодефициты, которые в большинстве случаев не наследуются.
Концепция первичных иммунодефицитных состояний сложилась в середине XX в. Однако клинические примеры данных патологий описывали и ранее.
Первичные иммунодефицитные состояния очень часто становятся причиной летального исхода пациентов моложе 20 лет. Основной причиной такового является присоединение вторичной инфекции. Дети до года, как правило, умирают из-за сепсиса, развившегося на фоне первичного иммунодефицита [1, 2].
В основе развития первичных иммунодефицитных состояний лежат мутация генов и перестройка хромосом. Ген, в котором есть дефект, является высоко пенетрантным при условии ранней экспрессии генетического дефекта [2–5].
Рассматриваемые иммунологические поражения можно получить с помощью генно-инженерных технологий нокаута генов. Это позволяет установить связь определенных генов с комплексом структур, которые они детерминируют, и процессами в иммунной системе. Рецессивные мутации, которые локализуются в Х-хромосоме, – одна из самых частых причин развития первичных иммунодефицитных состояний [6–9].
Х-сцепленное наследственное заболевание – болезнь Брутона, или первичный иммунодефицит гуморального звена иммунитета, впервые был описан в 1954 г. [2, 4, 5, 10–13]. Причиной его развития является дефект В-клеточной тирозинкиназы (Btk). Последняя участвует в формировании В-лимфоцитов. Как следствие, снижается уровень всех иммуноглобулинов (Ig). Важно отметить, что количество сывороточных иммуноглобулинов, указывающее на функцию В-клеток, а также на их взаимодействие с Т-хелперами, изменяется в большей степени, чем численность В-клеток.
Во время внутриутробного периода развития фиксируется снижение содержания материнских антител. Клинические признаки заболевания появляются в возрасте двух-трех лет. Чувствительность к токсинам и внеклеточным бактериям снижается. Реакция иммунитета на большинство вирусов не меняется. Исключение, в частности, составляют вирусы Коксаки и Экхо. Увеличения лимфоузлов не наблюдается. При пальпации они мягкоэластической консистенции, не спаяны с другими тканями, безболезненны. Размер печени и селезенки не увеличен. Часто встречаются респираторные аллергозы, отит, экзема, бактериальные конъюнктивиты, пиодермии. Из-за недостаточности секреторных IgA может развиться синдром мальабсорбции. Осложнениями заболевания являются ревматоидный артрит, остеомиелит, которые могут перейти в сепсис [1, 5, 10, 14, 15].
Диагноз подтверждается результатами лабораторных исследований: снижение количества В-лимфоцитов, иммуноглобулинов всех классов, в частности IgG, а также титром антител. Показатели Т-клеточного звена иммунитета остаются без изменений.
В качестве терапии назначают анибактериальные препараты, введение иммуноглобулинов (Иммуновенин, Пентаглобин, Сандоглобулин).
Прогноз, как правило, неблагоприятный.
Клинический случай
В республиканской детской клинической больнице г. Владикавказа в течение пяти лет наблюдался пациент с первичным иммунодефицитом гуморального звена иммунитета – болезнью Брутона. Помимо основного заболевания выявлены хронический рецидивирующий ювенильный артрит, пауциартикулярный вариант (активность первой степени, функциональное нарушение первой степени, рентгенологическое прогрессирование нулевой стадии), хронический гнойный средний отит, аденоиды третьей степени, анемия железодефицитная легкой степени.
Из анамнеза: родился от второй беременности, вторых срочных родов. Масса тела при рождении – 3 кг. Развивается в соответствии с возрастом. Перенес двустороннюю нижнедолевую пневмонию, правостороннюю верхнедолевую пневмонию (дважды), гнойный средний отит. Не привит.
В октябре 2014 г. поступил в хирургическое отделение республиканской детской клинической больницы с жалобами на отеки и боли в области голеностопных и коленных суставов, правого локтевого сустава, повышение температуры до 39 ºC. По результатам обследования поставлен диагноз: сепсис, септикопиемическая форма, острый гематогенный остеомиелит обеих бедренных костей, обеих большеберцовых костей, правой плечевой кости, гипохромная анемия средней степени.
В марте 2015 г. пациент неоднократно был госпитализирован в хирургическое отделение. Поставлен диагноз: хронический остеомиелит левого бедра, левосторонний гнойный гонит, двусторонний острый средний отит. Проведена антибактериальная терапия.
В апреле 2015 г. впервые обследован в отделении гематологии/ревматологии республиканской детской клинической больницы г. Владикавказа. Диагноз: ювенильный идиопатический артрит, серонегативный (активность второй степени, функциональное нарушение второй степени, рентгенологическое прогрессирование второй стадии). По результатам обследования поставлен диагноз: первичный иммунодефицит гуморального звена иммунитета – болезнь Брутона (агаммаглобулинемия), рецидивирующий гнойный средний правосторонний отит, хронический рецидивирующий ювенильный артрит, пауциартикулярный вариант (активность первой степени, функциональное нарушение первой степени, рентгенологическое прогрессирование нулевой стадии). Получал заместительную терапию внутривенными иммуноглобулинами (ВВИГ), нестероидные противовоспалительные препараты (Найз), метотрексат 5 мг один раз в неделю внутримышечно, преднизолон 15 мг с постепенной отменой, дипроспан в область коленного сустава и обоих голеностопных суставов. Через неделю после второго введения ВВИГ на фоне отмены глюкокортикостероидов зафиксировано выраженное обострение полиартрита с симметричным поражением суставов верхних и нижних конечностей. Повторно был назначен преднизолон, продолжено введение метотрексата.
В декабре 2015 г. в связи с персистированием артрита после снижения дозы и отмены глюкокортикостероидов назначен биологический препарат адалимумаб (Хумира) в дозе 40 мг один раз в две недели подкожно. Из-за повышения уровня трансаминаз, а также из-за рецидивирующего артрита метотрексат был отменен.
Последнее введение ВВИГ 18 января 2017 г. в отделении республиканской детской клинической больницы.
За прошедший период неоднократно переболел вирусными инфекциями, отмечались обострения отита, герпетические высыпания.
Из-за ухудшения состояния направлен участковым педиатром на обследование и лечение в республиканскую детскую клиническую больницу.
При поступлении обнаружена флегмона правой голени (в области икроножной мышцы), в связи с чем 1 июня 2017 г. проведена операция – вскрытие, санация, дренирование гнойника. С 1 по 9 июня 2017 г. находился на лечении в хирургическом отделении. Получал антибиотики, дезинтоксикационную терапию, ВВИГ (Иммуновенин) в дозе 10 г (2 и 3 июня).
Для дальнейшего обследования и лечения переведен в отделение гематологии/ревматологии.
В декабре 2018 г. ребенок направлен на стационарное лечение в республиканскую детскую клиническую больницу.
При поступлении масса тела – 15 кг, рост – 100 см. Нормального телосложения. Удовлетворительного питания.
Состояние средней тяжести. Кожные покровы бледно-розовые, единичные экхимозы на нижних конечностях. Периферические лимфоузлы единичные до 1 см. Дефигурация коленных суставов (в большей степени правого), умеренная болезненность при активных движениях.
Дыхание пуэрильное. Частота дыхания – 24 в минуту.
Область сердца на вид не изменена. Границы относительной сердечной тупости: правая – на 1 см снаружи от правого края грудины, левая – на 1 см снаружи от левой срединно-ключичной линии. Тоны сердца средней громкости, ритм не нарушен, короткий систолический шум на верхушке. Частота сердечных сокращений – 96 в минуту. Артериальное давление – 90/50 мм рт. ст.
Живот мягкий, безболезненный, печень и селезенка не увеличены. Стул и диурез не нарушены.
Результаты лабораторных анализов от 20 мая 2019 г. Общий анализ крови: уровень эритроцитов – 4,75 × 10 12 , гемоглобина – 110 г/л, тромбоцитов – 332 × 10 9 , ретикулоцитов – 13%, лейкоцитов – 8,5 × 10 9 , палочкоядерных нейтрофилов – 5%, эозинофилов – 9%, сегментоядерных нейтрофилов – 20%, лимфоцитов – 59%, моноцитов – 7%, скорость оседания эритроцитов – 4 мм/ч. Биохимический анализ крови: С-реактивный белок – отрицательно, IgA – 28 мг/дл (норма 80–406 мг/дл), IgM – 12 мг/дл (34–214 мг/дл), IgG – 813 мг/дл (680–1650 мг/дл), общий белок – 65 г/л, альбумин – 46 г/л, общий билирубин – 9,8 мкм/л, прямой – 1,9 мкм/л, сывороточное железо – 11,3 мкм/л, лактатдегидрогеназа – 872 ед/л, щелочная фосфатаза – 219 ед/л, мочевина – 4,8 мм/л, креатинин – 46 мкм/л, глюкоза – 4,4 мм/л, холестерин – 3,1 мм/л.
Общий анализ мочи от 20 мая 2019 г.: без патологии.
Результаты электрокардиограммы от 29 ноября 2018 г.: синусовый ритм, частота сердечных сокращений – 109 в минуту, горизонтальное положение электрической оси сердца.
Рентгенограмма органов грудной клетки от 21 мая 2019 г.: легочные поля прозрачны, тени корней не расширены, структура прослеживается, синусы свободные, тень средостения без особенностей.
Ультразвуковое исследование органов брюшной полости от 21 мая 2019 г.: печень на 1,5 см ниже реберной дуги, контур ровный четкий, эхоструктура паренхимы однородная, сосудистый рисунок сохранен, желчный пузырь 58 × 17 мм (норма 44 × 16 мм), деформирован, стенки тонкие, содержимое однородное, поджелудочная железа, селезенка, почки в норме.
Консультации врачей-специалистов: ЛОР от 28 мая 2019 г.: аденоиды третьей степени. Рекомендована аденотомия.
Исследован иммунный статус. Показатели клеточного иммунитета: CD3 – 50%, CD4 – 34,0%, CD8 – 27,0%, CD25 – 2,1%, соотношение CD4/CD8 – 1,24%, CD16 – 15,2%, CD18 – 68,3%, CD26 – 21,1%, CD45RA – 58,2%, CD95 – 35,2%, HLA-1 – 85,2%, HLA-DR – 10,3%, CD11b – 4,9%.
Факторы неспецифической резистентности: фагоцитарный индекс – 30%, фагоцитарное число – 2,8 ед., тест с нитросиним тетразолем – 6%, средний цитохимический индекс – 0,05, активность лизоцима – 20%, циркулирующие иммунные комплексы – 18 ед.
Рекомендовано: наблюдение педиатра, гематолога, иммунолога, ревматолога, отоларинголога. Аденотомия в плановом порядке. Проведение биохимического анализа крови один раз в три месяца плюс определение уровня аланинаминотрансферазы один раз в месяц. Внутривенное введение иммуноглобулина 10 г один раз в четыре недели. Подкожное введение препарата Хумира 40 мг один раз в две недели, метотрексата 5 мг один раз в неделю. Применение фолиевой кислоты 1 мг один раз в день, кроме дня введения метотрексата, Мальтофера в виде сиропа 10 мл один раз в день. Отвод от профилактических прививок.
Ребенок должен быть внесен в регистр страдающих орфанными заболеваниями.
В настоящий момент времени пациент находится на диспансерном наблюдении в поликлинике по месту жительства у педиатра, иммунолога и ревматолога. В случае необходимости может быть госпитализирован в республиканскую детскую клиническую больницу г. Владикавказа.
Болезнь Брутона – редкое заболевание, относящееся к группе первичных иммунодефицитных состояний. Недостаточная информированность педиатров о данной патологии приводит к несвоевременности диагностики и проведения адекватной терапии.
Приведенные нами данные могут способствовать повышению качества оказания медицинских услуг в условиях стационара и амбулаторно-поликлинического звена. Более ранняя диагностика и своевременно начатое лечение приведут к снижению частоты летальных исходов.
Агаммаглобулинемия, сцепленная с Х-хромосомой (синдром Брутона), характеризуется резким снижением иммуноглобулинов в сыворотке крови, клетки плазматического ряда не образуются, реакции клеточного иммунитета полностью сохранены.
Этиология: установлена наследственность, сцепленная с Х-хромосомой. В семьях заболевание проявляется в половине случаев. Возможны спонтанные мутации, наследственность в таких случаях не прослеживается. Патогенез не выяснен, полагают, что отсутствуют предшественники В-лим-фоцитов.
Бурсаэктомированные цыплята — экспериментальная модель этого заболевания. Клеточный иммунитет не нарушен. Характерны бактериальные инфекции, развивающиеся в раннем возрасте, либо до 1 года жизни, либо после 4—5 лет. Возникают повторные пневмонии с бронхоэктазами, отиты, менингит, сепсис.
Возбудители — грамположительные кокки. Вакцинация и вирусные инфекции протекают без особенностей. Диагностическое значение имеют лабораторные данные (резкое снижение IgG, отсутствие IgA и IgM). Антитела при иммунизации не возникают. Лимфопении не наблюдается.
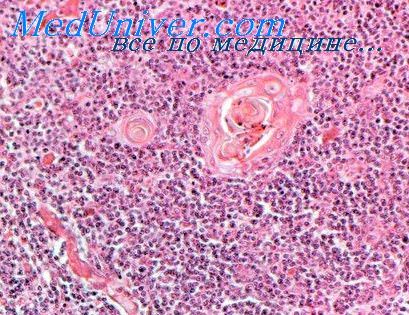
Типичными являются изменения лимфатических узлов, в которых не определяются фолликулы и кортикальная зона. Лимфатический узел сплошь представлен Т-зависимой паракортикальной зоной, в селезенке могут наблюдаться очень мелкие фолликулы, однако светлые центры отсутствуют.
Клетки плазматического ряда нигде не определяются. Особенно показательным является полное отсутствие плазматических клеток в слизистой оболочке кишки, которые в норме встречаются в большом количестве. Отсутствие плазматических клеток в костном мозге для детей до 6 лет не имеет диагностического значения.
Отсутствие плазматических клеток можно установить при жизни с помощью биопсии слизистой оболочки прямой кишки. Наряду с отсутствием плазматических клеток в слизистой оболочке часто обнаруживаются абсцессы со скоплением распадающихся лейкоцитов в криптах. В тимусе наблюдаются явления несвоевременного жирового метаморфоза с накоплением жира в клетках корковой зоны долек у детей даже в первые 6—7 мес жизни.
Типичным является развитие тяжелых бактериальных поражений дыхательного и пищеварительного тракта, пиодермии, гнойного менингита, сепсиса. Мы наблюдали тяжелый язвенно-некротический эзофагогастроэитероколит у мальчика 1 года 10 мес и кожный сепсис у мальчика 4 мес.
Наблюдается пневмоцистная пневмония без плазматических клеток в альвеолярных перегородках. Даже при систематическом лечении антибиотиками с заместительной терапией у-глобулином прогноз неблагоприятный. Смерть наступает в первые месяцы или первое десятилетие жизни. Описывается сочетание агаммаглобулинемии с лимфогранулематозом, злокачественной неходжкинской лимфой, аутоиммунными заболеваниями — ревматоидным артритом, дерматомиозитом и аутоиммунной гемолитической анемией.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
5.8. ИММУНОДЕФИЦИТНЫЕ СОСТОЯНИЯ
Иммунодефицитные состояния (ИДС) – это состояния, характеризую-щиеся снижением активности или неспособностью организма к эффективно-му осуществлению реакций клеточного и/или гуморального звена иммуните-та.
По происхождению все ИДС подразделяются на:
- первичные (наследственные, врожденные). Они являются результатом генетического дефекта, обусловливающего нарушения процессов про-лиферации, дифференцировки и функционирования клеток иммуноком-петентной системы;
- вторичные (приобретенные в постнатальном периоде). Развиваются под влиянием различных факторов физического или биологического харак-тера.
По преимущественному повреждению клеток иммунокомпетентной систе-мы различают 4 группы ИДС:
- с преимущественным повреждением клеточного иммунитета (Т-зависимые, клеточные);
- с преимущественным повреждением гуморального иммунитета (В-зависимые, гуморальные);
- с поражением системы фагоцитоза (А-зависимые);
- комбинированные, с поражением клеточного и гуморального звеньев иммунитета (Стефани Д.С.,ВельтищевЮ.Е., 1996; Ледванов М. Ю. , 1997; Резник И.Б.,1998; Хаитов Р.М.,Пинегин Б.В.,1999).
Физиологические ИДС включают в себя иммунодефициты (ИД) новорож-денных, беременных и лиц старческого возраста.
1. Иммунодефицит новорожденных. К моменту рождения у здоровых детей в крови содержатся материнские IgG и небольшое количество собст-венных IgG, IgM, IgA. Иммуноглобулины, полученные от матери, содержат антитела против всех видов микробов, с которыми контактировала мать, бла-годаря чему ребенок оказывается защищенным от них на протяжении первых месяцев жизни. Уровень материнских иммуноглобулинов постепенно снижа-ется, и максимальный дефицит их наблюдается через 2-3 месяца после рож-дения. Затем уровень собственных иммуноглобулинов ребенка в крови начи-нает постепенно повышаться, и количество IgM достигает нормального уровня взрослого человека в конце 1-го года жизни у мальчиков и 2-го – у девочек; IgG1 и IgG4 в возрасте 6-8 лет; IgG3 – в 10, а IgG2 - в 12 лет. Кон-центрация IgE достигает нормального уровня взрослого лишь спустя 10-15 лет после рождения. Секреторные IgA отсутствуют у новорожденных и по-являются через 3 месяца после рождения. Оптимальная концентрация секре-торного IgA устанавливается в возрасте 2-4 лет. Плазменный уровень IgA достигает такового показателя у взрослых в 10-12 лет. ИД новорожденных обусловлен тем, что высокое содержание лимфоцитов в периферической крови у новорожденных сочетается с их низкой активностью. У новорожден-ных детей отмечаются также низкая фагоцитарная активность и опсонизи-рующая способность крови. Уровень комплемента у новорожденных снижен и достигает уровня взрослого человека к 3-6-му месяцу жизни.
2. Иммунодефицит беременных. Иммунный статус беременных отлича-ется снижением числа Т- и В-лимфоцитов. Одновременно отмечается повы-шение активности С3-комплемента, что объясняют влиянием плацентарных стероидов на его синтез в печени.
3. Иммунодефицит лиц старческого возраста. Недостаточность имму-нитета при старении проявляется в снижении активности его гуморального и клеточного звеньев. При старении уменьшается общее число лимфоцитов пе-риферической крови. Функциональная активность Т- и В-лимфоцитов при старении падает, снижается интенсивность образования антител в ответ на антигенную стимуляцию. В старческом возрасте в основном продуцируются антитела класса IgM, резко снижена продукция IgA, IgG, подавляется синтез IgE, в связи с чем ослабевает течение атопических аллергических реакций. По мере старения уменьшается фагоцитарная активность макрофагов, ней-трофилов, снижаются активность комплемента, лизоцима и бактерицидная активность сыворотки крови.
Первичные ИДС - это генетически обусловленная неспособность орга-низма реализовать то или иное звено иммунного ответа. Эндогенные, как правило, генетически обусловленные дефекты одного из компонентов им-мунной системы приводят к нарушению системы защиты организма и кли-нически выявляются как одна из форм первичного ИДС. Так как в нормаль-ном функционировании иммунной системы и иммунном ответе участвуют многие типы клеток и сотни молекул, в основе первичного иммунодефицита лежат многочисленные варианты дефектов. Научная группа ВОЗ в 1997 г. выделила более 70 идентифицированных генетических дефектов на различ-ных уровнях преобразования стволовых клеток в Т- и В-лимфоциты или по-следующих этапах их дифференцировки, лежащих в основе первичных ИДС.
В последнее время в связи с обнаружением молекулярных дефектов, со-ставляющих основу многих иммунодефицитов, и существенной вариабель-ностью клинической картины и тяжести их течения, возможностью их позд-ней манифестации, в том числе у взрослых, становится ясно, что первичные ИДС – это не столь редкое состояние, как это считалось до сих пор. Частота значительной части первичных ИДС составляет 1/25000 - 1/50000, хотя такие варианты врожденных иммунных дефектов, как селективный дефицит IgA, встречается с частотой 1/500 - 1/700 человек. По данным ряда авторов, не-достаточность В-системы лимфоцитов и гуморального звена иммунитета от-мечается у 50-75% из общего числа больных ИДС; в 20% случаев отмечается комбинированная недостаточность клеточного и гуморального иммунитета; в 10% - изолированная недостаточность клеточного иммунитета, в 18% - не-достаточность фагоцитоза и в 2% - недостаточность системы комплемента.
ИДС с преимущественным нарушением клеточного звена иммунитета
Патология клеточного звена иммунитета проявляется на различных этапах созревания Т-лимфоцитов - от стволовой клетки до развития их специализи-рованных субпопуляций (рис. 1).
При дефектах преимущественно клеточного звена иммунитета характер-ны частые инфекции дыхательных и мочевыводящих путей, упорные рас-стройства пищеварения, хронический генерализованный кандидоз кожи и слизистых оболочек полости рта, пищеварительного тракта. Кандидозное по-ражение может выявляться в первые месяцы жизни в виде стоматита, дерма-тита, реактивной гиперплазии аденоидной ткани миндалин, лимфатических узлов, отмечается высокая интенсивность кариеса, развиваются бронхоле-гочная патология, фурункулез, в слюне повышено содержание секреторных IgA. Cледует отметить, что CD8 Т-лимфоциты осуществляют иммунологиче-ский надзор за внутренней средой, обеспечивая, в частности, элиминацию клеток, подвергшихся онкогенной трансформации. В случае недостаточности Т-системы лимфоцитов возникает онкогенноопасная ситуация.
1. Синдром Ди Джорджи возникает при гипо- и аплазии вилочковой же-лезы и паращитовидных желез. Заболевание обусловлено нарушением эм-бриональной дифференцировки эпителия в области 3-го и 4-го глоточных карманов. Число лимфоцитов в периферической крови значительно снижено. Синтез гуморальных антител не нарушен, но отмечается дефект в дифферен-цировке стволовых клеток в Т-лимфоциты. У детей с синдромом Ди Джорд-жи не отторгаются кожные трансплантаты, отсутствуют реакции ГЗТ. Забо-левание проявляется в периоде новорожденности (гипокальциемия, судороги, признаки кандидомикоза, инфекции дыхательных и мочевыводящих путей, упорные расстройства пищеварения).
2. Лимфоцитарная дисгенезия (синдром Незелофа) – количественная и качественная недостаточность Т-системы в результате атрофии тимуса и лимфатических узлов. Характеризуется отсутствием клеточных реакций им-мунологической защиты при нормальном содержании иммуноглобулинов в плазме крови. Проявляется в первые недели и месяцы жизни. Отмечаются за-держка развития ребенка, затяжной септический процесс с гнойно-воспалительными очагами во внутренних органах и коже. В периферической крови отмечается крайне низкое содержание лимфоцитов; резко угнетена ре-акция бласттрансформации лимфоцитов; слабо выражена реакция ГЗТ. Со-держание иммуноглобулинов всех классов в периферической крови – в пре-делах нормы. Дети чаще погибают в первые месяцы жизни от сепсиса.
ИДС с преимущественным повреждением В-системы
Гуморальные иммунодефициты относят к наиболее распространенным формам первичных ИДС.
2. Общая вариабельная гипогаммаглобулинемия (ОВГ). Это гетероген-ная группа ИДС, развитие которых связано с нарушением способности В-лимфоцитов трансформироваться в плазмоциты на фоне антигенной стиму-ляции. Количество циркулирующих в крови В-лимфоцитов не отличается от нормы, однако имеет место нарушение их дифференцировки. Для больных характерны гиперплазия лимфатических узлов, лимфоидного глоточного кольца, иногда – увеличение размеров селезенки. У детей с ОВГ не формиру-ется специфический иммунитет после вакцинации; у них обнаруживается склонность к развитию рецидивирующих воспалительных процессов инфек-ционной природы (синуситы, отиты, хронические пневмонии, фарингиты, тонзиллиты и др.). У взрослых больных ОВГ часто развиваются восходящий холангит и желчнокаменная болезнь, иногда артриты, атрофический гастрит. Общее количество иммуноглобулинов, особенно IgG, снижено.
Недостаточность местного иммунитета у больных ОВГ не коре-лирует с концентрацией плазматического уровня IgA и, вероятно, связана с наруше-ниями синтеза секреторных иммуноглобулинов. У многих больных отмечена склонность к аутоиммунным процессам. Установлено, что у больных ОВГ резко активированы естественные клетки-киллеры, причем их активность в 5 раз превышает нормальные значения.
3. Селективный дефицит иммуноглобулинов. Возможно развитие ИДС с селективным нарушением синтеза IgG, IgA. В основе их формирования могут лежать как блокада развития отдельных субпопуляций В-лимфоцитов, так и, что бывает чаще, повышение активности CD8 популяции лимфоцитов.
Дефицит субклассов IgG. ИДС развивается при дефиците каждого из подклассов, но при исследовании общего содержания IgG в крови редко об-наруживаются отклонения от нормальных значений, чаще оно в норме или повышено. Так как созревание клонов В-лимфоцитов, секретирующих IgG2 и IgG4, происходит не ранее 2-го года жизни, у детей раннего возраста имеется физиологический дефицит данных субклассов. Дефицит IgG2 обнаруживает-ся у 50% больных первичным ИДС, очень часто при общей вариабельной ги-поглобулинемии и, как правило, у детей старшего возраста проявляется хро-нической пневмонией и синдромом мальабсорбции. Селективный дефицит IgG1 может быть компенсирован за счет образования антител, относящихся к другим субклассам.
Изолированный дефицит IgA - одна из самых частых аномалий иммун-ной системы. Для него характерны низкое содержание IgA в сыворотке крови (менее 50 мг/л), отсутствие дефицита других классов иммуноглобулинов, нормальная способность организма к продукции антител, мало измененные показатели клеточного иммунитета. Так как IgA - основной иммуноглобулин системы местного иммунитета (секреторный IgA), обращают внимание на связь его дефицита с рецидивирующими и хроническими заболеваниями ды-хательных путей и ЛОР-органов. При отсутствии или низком содержании IgA в секретах создаются условия для развития аллергических и аутоиммун-ных заболеваний, предпосылки для развития дисбактериоза и воспалитель-ных заболеваний желудочно-кишечного тракта. С селективным дефицитом IgA может быть связано возникновение рецидивирующего герпетического стоматита, язвенного колита, регионального энтерита и др.
ИДС с поражением системы фагоцитоза
По механизму развития фагоцитарная недостаточность делится на три ос-новные формы:
Лейкопеническая - развивается вследствие подавления процессов проли-ферации и созревания моноцитов (ионизирующее излучение, ряд токсинов, цитостатики и др.) либо в результате наследственной блокады деления и дифференцировки, например миелоидной стволовой клетки.
Дисфункциональная - характеризуется расстройствами различных этапов процессов фагоцитоза и презентации антигена (подвижности фагоцитов, их адгезивных свойств, поглощения объекта фагоцитоза, переработки его и представления антигена лимфоцитам).
Дисрегуляторная - развивается вследствие нарушения регуляции различ-ных этапов фагоцитарной реакции биологически активными веществами (нейромедиаторами, гормонами, простагландинами, биогенными аминами, пептидами и др.).
Характеризуются нарушением дифференцировки стволовых клеток, бло-ком созревания Т- и В-лимфоцитов и их дефицитом. Комбинированные фор-мы иммунодефицита встречаются чаще, чем селективные. Как правило, они связаны с нарушением центральных органов иммунной системы. При комби-нированных ИДС ведущая роль принадлежит дефекту Т-клеток.
1. Cиндром ретикулярной дисгенезии - характеризуется уменьшением в костном мозге количества стволовых клеток. Характерна внутриутробная ги-бель плода, или дети гибнут вскоре после рождения.
Заболевание проявляется в первые месяцы жизни и часто характеризуется злокачественным течением. Наблюдается задержка прибавки массы тела, уже в первые дни жизни у некоторых детей появляются кореподобные высыпа-ния на коже, что может быть связано с реакциями несовместимости по отно-шению к материнским лимфоцитам, поступающим через плаценту в крово-ток ребенка. Развиваются признаки кожного кандидоза, диарея, острая ин-терстициальная пневмония, приобретающая затяжной и рецидивирующий характер. Дети очень восприимчивы к вирусным инфекциям. В крови выяв-ляется значительная лимфопения, особенно низко содержание Т-лимфоцитов. Содержание иммуноглобулинов всех классов заметно снижено. Исключение составляют грудные дети с IgG, полученными от матери. Патог-номоничны изменения вилочковой железы, гипоплазия миндалин и лимфа-тических узлов. Возникает неспособность проявлять реакции гиперчувстви-тельности замедленного типа. Дети редко доживают до 2-летнего возраста.
3. Синдром атаксии-телеангиэктазии (синдром Луи-Бар) обусловлен дефектом созревания, снижением функции Т-лимфоцитов, уменьшением их числа в крови (особенно Т-хелперов), дефицитом иммуно-глобулинов (осо-бенно IgA, IgE, реже IgG). Синдром характеризуется сочетанием атаксии и других неврологических отклонений с телеангиэктатическими изменениями сосудов склер, лица. Поражение нервной системы проявляется симптомами выпадения функций мозжечка, подкорковых ганглиев, диэнцефальной облас-ти, пирамидной системы. В результате их поражений возникают нарушение походки, замедленность произвольных движений, гиперкинезы, вегетососу-дистая дистония. У многих отмечаются вялотекущие пневмонии, развивают-ся ателектазы, пневмосклероз и бронхоэктазы. При исследовании лимфати-ческой системы устанавливается гипоплазия вилочковой железы, лимфатиче-ских узлов, селезенки. У многих детей отмечается уменьшение содержания в крови лимфоцитов, снижена реакция бласттрансформации лимфоцитов, не определяется IgA.
Заболевание характеризуется аутосомнорецессивным типом насле-дования.Прогноз синдрома неблагоприятен. Около 50% летальных исходов обусловлено хроническим поражением бронхолегочной системы, около 20% - развитием злокачественных процессов, которые связывают с утратой функ-циональной активности тимусзависимых лимфоцитов, а в общем плане – с отсутствием цензорной функции вилочковой железы – функции иммуноло-гического надзора. Некоторые больные доживают до 40-50 лет.
4. Синдром Вискотта-Олдрича характеризуется дефицитом перифери-ческих Т-лимфоцитов, нарушением их структуры и физико-химических свойств мембран, уменьшением клеточного иммунитета. Дефект, вероятно, связан с отсутствием на клеточной мембране лимфоцитов и тромбоцитов гликопротеида с относительной молекулярной массой 110 kD. При этом за-болевании выявляются снижение плотности микроворсинок на лимфоцитах, нарушение активации Т-лимфоцитов и не объясненная до сих пор нестабиль-ность экспрессии сиалогликопротеинов. Иммунологическая недостаточность обусловлена гипофункцией вилочковой железы. Часто обнаруживаются де-фицит IgM при нормальном содержании IgG и увеличение уровня IgA, IgE в крови. Характерно уменьшение продукции антител к антигенам-полисахаридам, но эти больные нормально реагируют на белковые антигены. Кроме того, имеют место врожденные дефекты тромбоцитов (нарушения ад-гезии, агрегации), тромбоцитопения. Болеют только мальчики. Заболевание проявляется в раннем возрасте, иногда в периоде новорожденности. Дети страдают частыми вирусными и бактериальными инфекциями. Характерны рецидивирующий гнойный отит, экзема, пиодермии, пневмония, колиты. Ге-моррагический синдром может быть ведущим.
Прогноз тяжелых форм неблагоприятен, дети погибают в возрасте до 10 лет. К летальному исходу приводят инфекции, геморрагии или злокачест-веннные новообразования лимфоретикулярной системы.
Недостаточность системы комплемента
Система комплемента представлена протеолитическими ферментами и ре-гуляторными белками. В крови имеются 20 комплементарных факторов, ак-тивация которых может осуществляться классическим или альтернативным путем. Активация комплемента обеспечивает защиту организма от любых чужеродных агентов; с активацией комплемента связаны и повреждающие эффекты при развитии аллергических и аутоиммунных реакций. При врож-денном и приобретенном расстройстве комплементарных факторов наруша-ются процессы фагоцитоза и происходит освобождение биологически актив-ных веществ.
При врожденном дефиците С1 невозможна активация системы компле-мента по классическому пути, поэтому, вследствие нарушения фаго-цитоза и лизиса микробов, наблюдаются повторные тяжелые гнойные процессы. При врожденном дефиците ингибитора С3б постоянно активируется комплемент С3, в результате чего содержание его в крови уменьшается. Хотя количество предшествующих комплементарных факторов (С1, С2, С4) не изменяется, однако из-за дефицита С3 нарушаются процессы фагоцитоза и лизиса бакте-рий, что проявляется повторными гнойными инфекциями.
При врожденном дефиците С5 склонность к инфекции также связана с на-рушением фагоцитоза и лизиса из-за невозможности образования соответст-вующих компонентов комплемента.
Содержание комплемента у детей на 30% ниже, чем у взрослых, что дела-ет понятным их склонность к инфекции и сепсису. Расстройства системы комплемента приобретенного характера проявляются в измене-нии количест-ва комплементарных факторов. При поражениях печени (цирроз, гепатит, хронический холецистит) нарушается синтез С1, С3, С6, С9. С другой сторо-ны, при аллергических, аутоиммунных заболеваниях комплементарные фак-торы уменьшаются в крови из-за связывания их иммунными комплексами.
Принципы лечения первичных ИДС
Лечение зависит от типа первичной иммунологической недостаточ-ности. Выделяются 3 основных направления иммунокоррекции.
Иммунная инженерия (трансплантация органов и тканей иммунной сис-темы: эмбриональной печени, тимуса, комплекса тимус-грудина, костного мозга, клеток иммунной системы; введение -глобулинов, иммуноглобулинов отдельных классов; сорбционные методы: гемосорбция, аффинная сорбция, иммуносорбция).
Коррекция гормонами и медиаторами иммунной системы (тимические гормональные факторы, миелопептиды, цитокины типа интерферона, интер-лейкины).
Фармакологическая коррекция - левамизол, диуцифон, полианионы и др.
Также применяется активная иммунизация против частых инфекций с по-мощью убитых вакцин, вводятся антибиотики, сульфаниламиды, противо-грибковые препараты.
Читайте также: