
Аутосомно рецессивные болезни реферат
Обновлено: 02.07.2024
Данная брошюра содержит информацию о том, что такое рецессивный тип наследования, и каким образом наследуются рецессивные заболевания. Для того, чтобы лучше понять особенности рецессивного наследования, вначале будет полезно узнать, что такое гены и хромосомы.
Что такое гены и хромосомы?
Наше тело состоит из миллионов клеток. Большинство клеток содержат полный набор генов. У человека тысячи генов. Гены можно сравнить с инструкциями, которые используются для контроля роста и согласованной работы всего организма. Гены отвечают за множество признаков нашего организма, например, за цвет глаз, группу крови или рост.
Гены расположены на нитевидных структурах, называемых хромосомами. В норме, в большинстве клеток организма содержится по 46 хромосом. Хромосомы передаются нам от родителей – 23 от мамы, и 23 от папы, поэтому мы часто похожи на своих родителей. Таким образом, у нас два набора по 23 хромосомы, или 23 пары хромосом. Так как на хромосомах расположены гены, мы наследуем по две копии каждого гена, по одной копии от каждого из родителей. Хромосомы (следовательно, и гены) состоят из химического соединения, называемого ДНК.
Иногда в одной копии гена возникает изменение (мутация), которое нарушает нормальную работу гена. Если такая мутация происходит только в одной копии гена рецессивного заболевания, а вторая копия нормальная, то обычно это не приводит к развитию генетического (наследственного) заболевания.
Рисунок 1: Гены, хромосомы и ДНК

Что такое аутосомно-рецессивное наследование?
Как наследуются рецессивные заболевания?
Рисунок 2: Как рецессивные заболевания передаются от родителя к ребенку

Если оба родителя являются носителями мутантной копии одного и того же гена, они могут предать своему ребенку как нормальную копию, так и измененную. Выбор происходит случайным образом.
Таким образом, каждый ребенок у родителей, которые являются носителями мутаций в одном и том же гене, имеет шанс, оцениваемый в 25% (1 из 4) унаследовать измененные копии гена от обоих родителей и, следовательно, оказаться больным. В то же самое время это означает, что существует шанс, оцениваемый в 75% (3 из 4), что ребенок не будет болен данным заболеванием. Оценка вероятности (25% или 75%) одна и та же для каждой новой беременности, и одинакова для мальчиков и для девочек.
Шанс унаследовать от родителей только одну копию измененного гена оценивается в 50% (2 из 4). Если такое происходит, ребенок будет здоровым носителем, как и его родители.
Наконец, существует шанс в 25% (1 из 4), что ребенок унаследует две нормальные копии гена, по одной от каждого из родителей. В этом случае у ребенка не будет заболевания, и он не будет являться носителем.
Процент риска остается одним и тем же при каждой беременности и одинаков как для мальчиков, так и для девочек.
Что происходит в том случае, если ребенок является первым в семье, у кого выявлено данное заболевание?
Иногда ребенок с рецессивным наследственным заболеванием может оказаться первым больным в семье. Несмотря на то, что во многих поколениях родственники могли быть носителями измененного гена, ребенок может оказаться единственным больным в семье, так как оба его родителя являются носителями, и он унаследовал измененные копии гена от обоих родителей.
Тест на определение носительства и пренатальная диагностика (тест во время беременности)
Для людей, у которых есть семейный анамнез рецессивного наследственного заболевания, существует несколько возможностей для обследования. Анализ на выявление носительства может быть проведен парам для выяснения, являются ли оба партнера носителями мутаций в определенном гене. Эта информация может оказаться полезной при планировании беременности. Для некоторых рецессивных заболеваний возможно проведение пренатальной диагностики (во время беременности) для определения, унаследовал ли будущий ребенок заболевание. Данная информация подробно обсуждается в брошюрах "Биопсия ворсин хориона" и "Аминиоцентез".
Другие члены семьи
Если кто-то в Вашей семье болен рецессивным заболеванием или является носителем, возможно, Вы захотите обсудить это с другими членами Вашей семьи. Это предоставит возможность Вашим родственникам, при желании, пройти обследование (специальный анализ крови) для выявления, является ли человек также носителем. Эта информация также может быть важна для родственников при диагностике заболевания. Это может быть особенно важно для тех родственников, у которых есть или будут дети. Подробно данная информация приведена в брощюре "Тестирование носителей"
Некоторым людям может оказаться сложно обсуждать свое генетическое заболевание с другими членам семьи. Они могут бояться причинить беспокойство членам семьи. В некоторых семьях люди из-за этого испытывают сложности в общении и теряют взаимопонимание с родственниками. Врачи-генетики, как правило, имеют большой опыт в решении подобных семейных ситуаций и могут помочь Вам в обсуждении проблемы с другими членами семьи.
Зависимость типа наследования генного заболевания от локализации патологической мутации на аутосоме. Болезни с аутосомно-доминантном рецессивным и Х - сцепленным типом наследования. Возможные варианты генотипов у потомков при таких заболеваниях.
| Рубрика | Медицина |
| Вид | реферат |
| Язык | русский |
| Дата добавления | 23.07.2015 |
| Размер файла | 31,4 K |
Студенты, аспиранты, молодые ученые, использующие базу знаний в своей учебе и работе, будут вам очень благодарны.
ТИПЫ НАСЛЕДОВАНИЯ
Общепринятой классификации генных болезней не существует. Исходя из генетического принципа, выделяют генные болезни с аутосомно- доминантным, аутосомно-рецессивным, Х - сцепленным рецессивным, Х - сцепленным доминантным, У-сцепленным (голандрическим) и митохондриальным (цитоплазматическим) типами наследования.
Тип наследования генного заболевания зависит от локализации патологической мутации на аутосоме или половой хромосоме, от ее доминантности или рецессивности, а также оттого, где она возникла: в ядерной ДНК или митохондриальной.
1. Аутосомно-доминантный тип наследования
Примерами заболевания с аутосомно-доминантным типом наследования могут служить синдромы Ваарденбурга, Марфана, Маршалла, Стиклера, нейрофиброматоз Реклингаузена, нижнечелюстной дизостоз.
Болезни с аутосомно-доминантным типом наследования
При аутосомно-доминантных заболеваниях только браки Аа х аа имеют практическое значение. Гомозиготный по патологическому доминантному гену генотип больного родителя (брак АА х аа) возможен только при ассортативности (избирательности, неслучайности) браков в популяции. Такие браки маловероятны из-за, относительно редкой встречаемости и сниженной репродуктивной способности больных с врожденными или выявляющимися в детстве болезнями с аутосомно-доминантным типом наследования. В литературе имеются данные о возможности изменения и увеличении тяжести заболевания при гомозиготизации аутосомно - доминантных генов. Так, фогель и Мотулский (1989) приводит случай, олигодактилии у ребенка (АА) ,от брака двоюродных сибсов с брахидактилией(Аа хАа).
Если, один из родителей гетерозиготен по аутосомно-доминантному гену (Аа), а другой гомозиготен по нормальному аллелю (аа), т,о в таком браке возможны следующие варианты генотипов у потомков:
Родители Аа х аа
Гаметы А А а а
Дети Аа; Аа; аа; аа
Таким образом, каждый ребенок в браке больного (Аа) со здоровым (аа) имеет 50%-ную вероятность (риск) получить нормальный аллель (а) и быть здоровым, а также вероятность (риск) унаследовать патологическую мутацию (А) и быть больным. При этом соотношение здоровых и больных детей в потомстве составляет 1 : 1 и не зависит от пола ребенка.
Для болезней с аутосомно-доминантным типом наследования характерна встречаемость патологии у гетерозиготных носителей (Аа) в каждом поколении и соотношение 1 : 1 больных и здоровых среди сибсов. Для генных болезней с данным типом наследования типичны неполная пенетрантность (проявляемость) и варьирующая экспрессивность (выраженность). Последняя затрагивает не только разные семьи, нои членов одной и той же семьи, затрудняя диагностику. Заболевания с аутосомно -доминантным типом наследования могут быть как врожденными, так и выявляться в любом возрасте. Например, хорея Гентингтона и болезнь Альцгеймера проявляются в возрасте 35-40 лет и после 60 соответственно. Наконец, аутосомно-доминантным болезням свойственно протекание с повышенной тяжестью и даже изменением фенотипа у гомозиготных доминантных индивидов.
Примеры некоторых аутосомно- доминантных болезней приведены в таблице 1.
Таблица 1. Болезни с аутосомно-рецессивным типом наследования
7q31.2
концентрация ионов натрия
в потовой жидкости, повышение функции
1q13 и 14q22
Короткая шея, отечные
рот, макроглоссия, сухая
кожа, редкие волосы,
йода в щитовидной
железе при нормальном
Необычный запах мочи
и тела, пониженная
волос, глаз, микроцефалия,
ного развития, симптом
на глазном дне, децереб-
Отставание в психомоторном-
форма 16q22
подвывих хрусталика, миопия,
Умеренное отставание в
развития, роста, помутнение
роговицы, черепно- лицевые
переносица, большие щеки,
Практически у всех больных, страдающих патологией с аутосомно-доминантным типом наследования, отмечается нарушение репродуктивной функции, а иногда и стерильность, что может быть связано как с биологическими, так и социальными факторами. Так, новыми мутациями обусловлено 80-90% всех случаев ахондроплазии, 30-50% случаев нейрофиброматоза-1. Исключением из этого правила являются болезни с поздним началом, когда к началу болезни деторождение уже закончено. Для родителей ребенка с новой мутацией, возникшей в половой клетке одного из них, повторный риск рождения больного ребенка не превышает популяционный, а для самого ребенка равен 50% . Вероятность возникновения доминантной мутации в половой клетке у пожилых отцов выше, чем у молодых.
Для распознавания аутосомно-доминантного типа наследования наиболее важными являются следующие признаки:
. признак (болезнь) проявляется в каждом поколении без пропусков (вертикальный тип наследования), исключая случаи неполной пенетрантности (проявляемости) гена;
. любой ребенок больного с аутосомно-доминантным заболеванием имеет 50% -ный риск унаследовать это заболевание от родителей (в случае полной пенетрантности);
. непораженные члены семьи (для заболеваний со 100% -ной пенетрантностью) не могут иметь больных детей;
. встречаемость и передача аутосомно- доминантных заболеваний не связана с полом, т.е. лица мужского и женского пола поражаются одинаково часто.
2.Болезни с аутосомно-рецессивным типом наследования
Аутосомно-рецессивные заболевания проявляются только у гомозигот, которые получают по одному рецессивному гену от каждого из родителей. Заболевание может повторяться у сибсов пробанда, распространяясь вширь в пределах одного поколения (горизонтальный тип наследования). Характерным типом брака при аутосомно-рецессивных заболеваниях является брак (Аа хАа): родители здоровы, но являются носителями патологического гена. В таком браке вероятность рождения больного ребенка составляет 25%.
Родители Аа х Аа
Гаметы А а А а
Дети АА; Аа; Аа; аа
В связи с тем, что больные дети рождаются у здоровых родителей, такие семьи можно выявить только после рождения больного ребенка, ретроспективно установить генотипы родителей и повторный риск рождения больного ребенка. Носители аутосомно-рецессивного гена - редкое явление, поэтому их случайная встреча маловероятна. Напротив, при кровно - родственных браках вероятность такой встречи повышается, так как оба супруга могут унаследовать редкий рецессивный ген от общего порядка. Если, например, супруги являются двоюродными братом и сестрой, то такой ген они могут унаследовать от бабки или деда.
По аутосомно-рецессивному типу наследуются подавляющее большинство врожденных нарушений обмена, муковисцидоз, синдромы Лоуренса-Муна и Барде-Бидля и другие, всeгo 70 нозологических единиц. На сегодняшний день основными методами их предупреждения являются медико-генетическое консультирование и пренатальная (дородовая) диагностика в тех случаях, когда такие методы разработаны. Важное значение имеет возможность выявления гетерозиготных носителей.
Браки Аа х аа встречаются редко. Встреча двух супругов с такими генотипами более вероятна, если брак кровно - родственный. Возможны такие случаи, когда больной (аа) супруг выбирает партнера из семьи с аналогичным страданием, например глухотой. Характер расщепления потомства при таких браках имитирует аутосомно-доминантный тип наследования.
Родители Аа х аа
Гаметы А а а а
Дети Аа; Аа; аа; аа
В некоторых случаях больные дети с аутосомно-рецессивной патологией рождаются также в браках аа х аа. В таком типе брака вероятность рождения больного ребенка составляет 100%.
Краткая характеристика аутосомно-рецессивного типа наследования включает:
. заболевания прослеживаются в родословной по горизонтали (чаще в пределах одного поколения), в основном среди сибсов пробанда;
. повторный риск рождения больного ребенка у здоровых родителей составляет 25%;
. отмечается повышенная частота кровно - родственных браков среди родителей пробандов;
. оба пола поражаются с одинаковый частотой.
3. Х-сцепленное наследование
Локализованные на половых хромосомах гены носят название сцепленных с полом. Сцепленные с полом гены могут располагаться как на Х-хромосоме, так и на У - хромосоме. Однако в клинической генетике практическое значение имеют Х - сцепленные заболевания, т.е. такие, при которых патологические гены располагаются на Х-хромосоме.
Распределение Х - сцепленного признака в потомстве зависит от распределения Х -хромосомы, несущей аномальный ген. Поскольку у женщин имеются две Х -хромосомы, а у мужчин одна, то возможны следующие варианты генотипов: у мужчин - ХАУ; ХаУ, у женщин - ХАХА; ХАХа; ХаХа; (ХА - доминантный ген, расположенный на Х-хромосоме, Ха - рецессивный ген, расположенный на Х-хромосоме).
Таким образом, у женщин возможны: гомозиготный по доминантному аллелю генотип, гетерозиготный генотип и гомозиготный по рецессивному аллелю генотип. У мужчин возможен только гемизиготный генотип, т.к. аллель, расположенный на Х-хромосоме, у мужчины не имеет пары на У-хромосоме.
Х - сцепленное, рецессивное наследование
Х - сцепленные рецессивные заболевания проявляются у мужчин, имеющих соответствующий ген, а у женщин только в случае гомозиготного состояния (что наблюдается крайне редко), чаще при кровно - родственных браках.
Используя приведенные выше обозначения, можно определить все возможные генотипы детей в потомстве больного мужчины и здоровой женщины:
Родители ХаУ х ХАХА
Гаметы Ха У ХА ХА
Дети ХАХа; ХАХа; ХАУ; ХАУ
Согласно схеме все дети будут фенотипически здоровы, но генотипически все дочери являются гетерозиготными носителями. Если женщина-носитель выйдет замуж за здорового мужчину, возможны следующие варианты в потомстве:
Родители ХАУ х ХАХа
Гаметы ХА У ХА Ха
Дети ХАХА; ХАХа; ХАУ; ХаУ
Дочери в 50% случаев будут носительницами патологического гена, а для сыновей существует 50% -ный риск быть больными.
Таким образом, основные критерии болезней с Х - сцепленным типом наследования следующие:
1. Заболевание встречается в основном у лиц мужского пола. Больные гомозиготные женщины при Х - сцепленных рецессивных заболеваниях являются исключением, которое наблюдается в том случае, если больной мужчина вступает в брак с носительницей гена этого заболевания.
3. Заболевание никогда не передается от отца к сыну.
4. У носителей могут выявляться субклинические признаки заболевания.
5. Степень риска для сыновей женщины, являющейся достоверно носительницей заболевания, составляет 50%.
6. Половина дочерей женщины - носительницы заболевания также будут носительницами.
Все фенотипически здоровые дочери пораженного отца являются облигатными гетерозиготными носительницами.
Сама по себе передача признака от пораженных дедов через здоровых матерей пораженным внукам еще не может служить доказательством локализации гена в Х-хромосоме. Аналогичный тип передачи возможен и в случае аутосомного гена, проявление которого ограничено мужским полом. Решающим является тот факт, что все сыновья пораженных мужчин здоровы. Однако этим критерием невозможно воспользоваться, если заболевание настолько тяжелое, что больные не оставляют потомства.
В отличие от Х-сцепленного рецессивного наследования заболевания с Х-сцепленным доминантным наследованием встречаются в два раза чаще у женщин, чем у мужчин. Пораженные индивиды обычно имеют нормальную репродуктивную способность. Главная характеристика Х-сцепленного доминантного наследования заключается в том, что больные мужчины передают ген (или заболевание) всем своим дочерям и ни одному из сыновей. Больная женщина передает Х- сцепленный доминантный ген половине своих детей независимо от пола, как и при аутосомно-доминантном типе наследования. Таким образом, лишь дети пораженных отцов дают возможность различить Х- сцепленное доминантное и аутосомно-доминантное наследование. Для всех признаков с установленным Х-сцепленным доминантным типом наследования было показано, что в среднем мужчины поражены тяжелее, чем женщины. Это закономерно, поскольку у гетерозиготных женщин частичная компенсация может определяться наличием нормального аллеля в другой Х-хромосоме. Полностью это факт стал, объясним после открытия феномена случайной инактивации одной из К-хромосом у женщин (лайонизации). Х-сцепленное доминантное наследование происходит при летальности мужчин-гемизигот.
Как уже отмечалось, у женщин Х-сцепленные заболевания обычно имеют менее тяжелые проявления, чем у мужчин. В некоторых случаях поражение мужских зигот оказывается настолько тяжелым, что они погибают внутриутробно. Тогда в родословных среди пораженных должны быть только женщины, а среди их пораженных детей - только дочери, причем в соотношении со здоровыми дочерьми и сыновьями 1: 1: 1. Кроме того, мужские гемизиготы, которые не погибают на очень ранней стадии беременности, должны обнаруживаться в спонтанных абортах или среди мертворожденных мальчиков. Ленц (1961) первым показал, что этот тип наследования существует у человека для заболевания, известного под названием недержания пигмента (синдром Блоха-Сульцбергера). Предполагается, что летальность мужских плодов имеет место при рото - лице-пальцевом синдроме (множественные гиперплазированные уздечки языка, расщелины губы и нёба, гипоплазия крыльев носа, асимметричное укорочение пальцев), синдроме Ретта - Гольтца и других болезнях.
Примером Х - сцепленного рецессивного заболевания является гемофилия - А - несвертываемость крови вследствие дефицита восьмого фактора свертывающей системы крови. Клинические признаки включают частые и длительные кровотечения, даже из небольшой ранки, кровоизлияния во внутренние органы и суставы. Частота заболевания - 1 на 10000 новорожденных мальчиков. Гемофилией страдают, как правило, мужчины, причем матери последних здоровые женщины, как правило, носительницы рецессивного гена гемофилии. Если мужчины - гемофилики вступают в брак со здоровыми женщинами, то их сыновья унаследуют хромосому У, свободную от этого гена. Они здоровы и не носят гена гемофилии. Дочери мужчин -гемофиликов фенотипически здоровы, но все являются гетерозиготными по гену гемофилии, т.е. носительницами - этого гена. Их сыновья, в 50% случаев также унаследуют гены гемофилии и окажутся больными. Гетерозиготными окажутся и 50% дочерей такой матери. Поскольку у мальчиков нет второй Х-хромосомы, то рецессивный мутантный, ген гемофилии проявляет свое действие, и дети страдают гемофилией. У девочек две хромосомы Х, на второй хромосоме Х локализован доминантный (нормальный), ген, поэтому унаследованный рецессивный ген не обнаруживает своего действия - девочки не болеют гемофилией. Таким образом, в рассмотренном случае 50% мальчиков будут поражены гемофилией и 50% девочек окажутся гетерозиготными носительницами гемофилии.
Гемофилией могут страдать и женщины. В литературе описаны такие случаи, но они имеют место лишь тогда, когда девочки рождаются от родителей, один из которых является гемофиликом (отец), другой - гетерозиготным носителем (мать). Вероятность такoгo брака невелика.
Передача рецессивного гена, детерминирующего гемофилию, от гетерозиготных носительниц к их дочерям, внукам и т.д., которые становятся гетерозиготными носительницами и сыновья которых в 50% случаев болеют гемофилией, хорошо прослеживается при ознакомлении с генеалогией некоторых царствовавших семей в Европе. Их родословная идет от английской королевы Виктории, бывшей гетерозиготной по гену гемофилии. От гемофилии умерли три правнука королевы Виктории - испанские инфанты Альфонс, Гонзало - Джеймс, являвшиеся сыновьями Альфонса ХIII и Виктории Евгении Баттенбергской. Гемофиликом был также сын последнего русского царя Николая 11 Алексей, который унаследовал ген гемофилии от своей матери, царицы Александры Федоровны (Алисы), а последняя, в свою очередь, получила его через мать от своей прабабки- королевы Виктории.
Таблица 2. Выявление носителей при Х-сцепленных заболеваниях (по Ф. Фогель и А. Мотулъски, 1989 г.)
Аутосомно-рецессивные заболевания проявляются только у гомозигот или компаунд-гетерозигот, людей с двумя мутантными и отсутствием нормального аллеля, так как при этих болезнях одна нормальная копия гена компенсирует мутантный аллель и предохраняет от появления болезни.
Поскольку человек наследует от одного родителя только один из двух аллелей в любом локусе, гомозиготы должны унаследовать по мутантному аллелю от каждого родителя (исключая однородительскую дисомию или новые мутации, которые редко встречаются при аутосомно-рецессивных заболеваниях).
К гомозиготному потомству, пораженному аутосомно-рецессивной болезнью, могут приводить три типа браков. Мутантный рецессивный аллель обозначен буквой r, а нормальный доминантный — R. Хотя любой брак, когда оба родителя имеют хотя бы один рецессивный аллель, может приводить к появлению гомозиготного потомства, наиболее часто бывают браки между двумя простыми гетерозиготами.
Если оба родителя больного — гетерозиготы (носители), риск их детей получить рецессивный аллель от каждого родителя равен 1/2, и, таким образом, вероятность наследования двух рецессивных аллелей и, следовательно, болезни — 1/2x1/2=1/4. Пробанд может быть единственным пораженным членом семьи, а если есть другие больные, они обычно находятся в том же сибстве и отсутствуют в родословной.
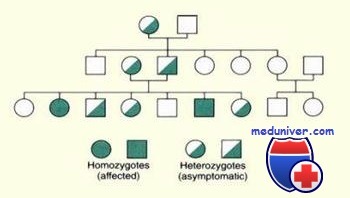
Аутосомно-рецессивное наследование
Опосредованные полом аутосомно-рецессивные болезни
Поскольку мужчины и женщины имеют одинаковый комплект аутосом, аутосомно-рецессивные болезни обычно встречаются с одинаковой частотой и тяжестью течения у мужчин и женщин. Тем не менее существуют и исключения. Некоторые аутосомно-рецессивные фенотипы зависят от пола, т.е. проявляются у обоих полов, но с разными частотами или тяжестью.
Примером фенотипа аутосомной болезни, чаще встречающейся у мужчин, служит гемохроматоз. Это аутосомно-рецессивное заболевание метаболизма железа наиболее часто, приблизительно у 0,5% населения, встречающееся в Северной Европе, вызвано гомозиготной нонсенс-мутацией в ген HFE, заменяющей цистеин тирозином в 282 положении (Cys282Tyr).
У гомозигот по мутации Cys282Tyr усилено всасывание пищевого железа и у них часто обнаруживают отклонения в лабораторных данных, напоминающие чрезмерное накопление тканевого железа, хотя заболевание лишь изредка приводит к реальному избытку железа с серьезным поражением сердца, печени и поджелудочной железы.
Более низкая встречаемость клинических проявлений заболевания у женщин (1/5 или даже 1/10 числа мужчин) обусловлена, среди других факторов, уменьшенным поступлением железа с пищей, меньшим употреблением спиртных напитков и повышенной потерей железа при менструациях.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021

Наследственные заболевания передаются от одного или обоих родителей детям. Они вызываются генетическими мутациями, но далеко не все генетические заболевания являются наследственными. Как в этом разобраться, какие виды заболеваний бывают, как их лечить и как диагностировать — рассказываем в нашей статье.
Содержание
Что такое наследственные заболевания?
Наследственные заболевания — это заболевания, обусловленные генными или хромосомными мутациями. У людей от 20 000 до 25 000 генов. Генетическая мутация возникает, когда изменяется один или несколько генов. Если это генетическое изменение передается детям, то это наследственное генетическое заболевание.
При совпадении у партнеров статусов носительства определенных болезней есть высокий риск рождения ребенка с наследственным заболеванием. Если у вас не проявляются симптомы заболевания, вы по-прежнему можете быть носителем и передать мутации своим детям.
Многие генетически обусловленные заболевания проявляются не сразу после рождения, а спустя некоторое время. От наследственных заболеваний следует отличать врожденные заболевания, вызванные внутриутробными повреждениями, например, инфекцией или внешними воздействиями.
Чем отличаются наследственные заболевания от врожденных нарушений?
Генетические заболевания являются результатом изменения одного или нескольких генов и могут передаваться в поколениях или нет.
Все наследственные заболевания имеют генетическое происхождение, т. е. являются результатом изменения одного или нескольких генов и передаются из поколения в поколение. Симптомы могут не проявляться с самого рождения.
Врожденные нарушения могут быть наследственными или нет, а симптомы могут проявляться с рождения. Но их появление не обязательно связано с генетикой.
Виды наследственных заболеваний
Наследственные заболевания разделяются на хромосомные, генные и митохондриальные.
Хромосомные заболевания
В настоящее время описано около 1000 форм хромосомных заболеваний. Хромосомные заболевания возникают в результате изменения числа или структуры хромосом. Они характеризуются общими признаками: маленькая масса и длина тела при рождении, отставание в умственном и физическом развитии, задержка и аномалии полового развития и прочее.
Моногенные заболевания
Моногенные заболевания возникают в результате повреждения ДНК на уровне гена. Количество моногенных заболеваний по некоторым оценкам достигает 5000.
Среди признаков моногенных болезней можно выделить: различные формы умственной отсталости, дефекты органов слуха, зрения, скелетные дисплазии, болезни нервной, эндокринной, иммунной и других систем. К числу наиболее известных моногенных болезней относятся муковисцидоз, гемофилия А и В, болезнь Гоше, миодистрофия Дюшенна/Беккера, спинальная мышечная атрофия, дальтонизм.
Выявить тяжелые моногенные заболевания можно с помощью пренатальной диагностики, а также, определив наличие мутаций у родителей с помощью генетического теста.
Интереснее всего мне было узнать об особенностях метаболизма. Именно поэтому я выбрала Атлас: только тут есть достаточно объемный раздел на эту тему. Например, всю жизнь я борюсь с весом, мигренью, болями в шее и спине, анемией.
Митохондриальные заболевания
Митохондриальные заболевания обусловлены генетическими, структурными, биохимическими дефектами в функционировании митохондрий, которые приводят к нарушению тканевого дыхания.
Митохондрии содержат свою собственную ДНК. А болезни, вызванные мутациями в митохондриальной ДНК, наследуются исключительно по материнской линии. Если именно таким образом было унаследовано митохондриальное заболевание, существует 100% вероятность того, что каждый ребенок в семье его унаследует.
Симптомы могут включать в себя: нарушение роста, слабость мышц, аутизм, ментальные расстройства, проблемы с дыханием, слухом и зрением. Примеры митохондриальных заболеваний: синдром Лея, синдром Вольфа-Паркинсона-Уайта, наследственная оптическая нейропатия Лебера и другие.
Полигенные или мультифакториальные заболевания
Существуют также болезни с наследственной предрасположенностью, которые называют мультифакториальными или полигенными заболеваниями.
Мультифакториальные заболевания обусловлены наследственными факторами риска, и в значительной степени — неблагоприятным воздействием среды. К мультифакториальным заболеваниям относятся большинство хронических заболеваний, включая сердечно-сосудистые, эндокринные, иммунные, нервно-психические, онкологические и др. Например, бронхиальная астма, сахарный диабет, ревматоидный артрит, гипертоническая болезнь сердца и т.д.
Как передаются наследственные заболевания?
Организм человека состоит из триллионов клеток. Каждая клетка имеет ядро, которое содержит хромосомы. Каждая хромосома состоит из плотно свернутых нитей дезоксирибонуклеиновой кислоты (ДНК).
Гены — это инструкции по сборке белков в нашем организме, которые определяют специфические черты каждого человека, например, цвет глаз или волос. Большинство клеток в организме обычно содержат 46 хромосом, организованных в 23 пары. В каждой из этих 23 пар есть одна унаследованная хромосома от отца и одна — от матери. Из 23 пар 22 пары одинаковые у женских и мужских организмов, а одна оставшаяся определяет, являетесь вы мужчиной (XY) или женщиной (XX).
Мутации, из-за которых возникают наследственные заболевания, могут иметь доминантный или рецессивный характер наследования.
Доминантное наследование означает, что только одна копия гена — от матери или отца — должна иметь мутацию (или патогенный вариант гена) для проявления признака или заболевания. А при рецессивном типе человек наследует две измененные копии одного и того же гена.
Аутосомно-доминантный паттерн наследования
При аутосомно-доминантном наследовании заболеваний генетически обусловленная болезнь проявляется в том случае, если у человека есть хотя бы один мутированный ген, и этот ген не расположен на половых (Х и Y) хромосомах.
Болезнь Хантингтона и синдром Марфана — два примера аутосомно-доминантных болезней. Мутации в генах BRCA1 и BRCA2, которые также связаны с раком молочной железы, передаются по этой схеме.
Аутосомно-рецессивный паттерн наследования
При аутосомно-рецессивном наследовании мутируют обе копии генов. Чтобы унаследовать аутосомно — рецессивное заболевание, такое как муковисцидоз, спинальная мышечная атрофия, или фенилкетонурия (ФКУ), оба родителя должны быть носителями. Ребенок наследует две копии дефектного гена — по одной от каждого родителя. Например, люди, имеющие одну копию гена с мутацией, а вторую — без мутации, называются носителями, потому что сами они здоровы.
Х-сцепленное рецессивное наследование
В Х-сцепленном рецессивном наследовании мутированный ген находится на Х-хромосоме. Болезнь проявляется только в случае, если другой Х-хромосомы с нормальной копией того же гена у человека нет.
Мышечная дистрофия Дюшенна, некоторые виды дальтонизма и гемофилия А — примеры рецессивных заболеваний, связанных с X-хромосомой. Мужчина с рецессивным заболеванием, связанным с X-хромосомой, передаст свою нетронутую Y-хромосому сыновьям, и ни один из них не пострадает. Если он передаст свою Х-хромосому (с дефектным геном) своим дочерям, то все они будут носителями болезни. У его дочерей может не быть симптомов или только легкие признаки заболевания, но они могут передать мутированный ген своим детям.
Женщины-носители рецессивного заболевания, связанного с X-хромосомой, часто имеют лёгкие признаки заболевания или вообще не имеют симптомов. Это связано с тем, что у женщин-носителей есть одна нормальная копия гена и одна мутированная копия. Нормальная копия обычно компенсирует дефектную копию в женском организме, в отличие от мужчин, у которых только одна X-хромосома.
Женщины, имеющие только один патологический ген, передают заболевание в среднем половине своих детей вне зависимости от пола. Женщины же, имеющие два патологических гена, передают заболевание всем своим детям. К таким заболеваниям относятся гемофилия А и дальтонизм.

Как генетическое тестирование помогает при планировании семьи
Если вы знаете или предполагаете, что у вас или вашего партнера в семейной истории есть какое-либо генетическое заболевание, вы можете определить это с помощью Генетического теста Атлас. Генетическое консультирование поможет вам узнать о методах лечения, профилактических мерах и репродуктивных возможностях.
Как лечить наследственные заболевания и как с ними жить?
Раньше наследственные заболевания были неизлечимы. Сейчас это по-прежнему остаётся проблемой для многих заболеваний, но для некоторых из них методы лечения уже найдены. Например, это касается болезней, связанных с нарушением метаболизма.
При большинстве наследственных нарушений обмена веществ один фермент либо вообще не вырабатывается организмом, либо вырабатывается в форме, которая не работает. Например, при отсутствии какого-либо фермента в организме могут накапливаться токсичные вещества или может не синтезироваться необходимый продукт — как при гемохроматозе 1 типа.
При этом заболевании организм поглощает слишком много железа из пищи и не может естественным образом избавиться от избытка. Это может привести к чрезмерному накоплению железа в сердце, поджелудочной железе и печени.
Лечение генетических нарушений обмена веществ следует двум общим принципам:
- Необходимо сократить или исключить прием любой пищи или лекарств, которые не усваиваются организмом.
- Заменить или восполнить отсутствующий или неактивный фермент для восстановления метаболизма с помощью диеты и/или лекарств.
Есть более серьезные и распространенные наследственные заболевания, которые не лечатся. Например, мековисцидоз — скопление слизи в лёгких и в пищеварительной системе. От муковисцидоза нет лекарства, но разные методы контроля симптомов помогают предотвращать или уменьшать осложнения и облегчать жизнь с этим заболеванием.
Со временем муковисцидоз прогрессирует и может привести к летальному исходу, особенно при наличии сопутствующих инфекций. Сегодня благодаря достижениям медицины около половины людей с муковисцидозом доживают до 40 лет. Дети, рожденные с этим заболеванием в наши дни, смогут прожить ещё дольше.
Одно из самых тяжелых наследственных заболеваний, спинальная мышечная атрофия, также с недавнего времени поддается лечению с помощью генной терапии. Но доступен этот метод далеко не каждому. Препарат для лечения СМА — самый дорогой лекарственный препарат в мире.
Как я могу узнать, что являюсь носителем генетического заболевания?
Наши гены содержат инструкции, которые сообщают организму, как правильно функционировать. При изменении этих инструкций развиваются различные заболевания. Во многих случаях симптомы впервые проявляются в зрелом возрасте, поэтому иногда мы не знаем, что являемся носителями. Предупредить риски развития и передачи наследственного заболевания можно с помощью Генетического теста Атлас.

Наследственные заболевания — заболевания, этиологическим фактором которого является мутация. Хромосомные болезни — группа наследственных заболеваний, обусловленных изменением числа и структуры хромосом.
Частота хромосомных болезней составляет — 5-7 на 1000 новорожденных.
Различают следующие хромосомные нарушения:
1). Болезни, связанные с нарушением числа аутосом (анеуплоидии, триплоидии и др.).
2). Болезни, связанные с изменением числа хромосом (трисомиии, моносомии).
3). Болезни, связанные с изменением структуры хромосом (делеции, дупликации, инверсии, транс локации: сбалансированные и несбалансированные).
Наследственные заболевания, обусловленные микроделециями и микродупликациями хромосом (болезни геномного импритинга).
Моногенные болезни — заболевания, обусловленные мутациями на уровне гена.
Тип наследования — аутосомно доминантный, аутосомно-рецессивный, Х-сцепленный доминантный, Х-сцепленный рецессивный.
Митохондриальные заболевания — передаются от матери с митохондриальной ДНК.
Мультифакториальные заболевания, врожденные пороки развития.
В мире описано более 300 наследственных заболеваний с нарушением слуха, различными формами тугоухости, пороками развития ушной раковины.
В статье описаны наиболее часто встречающиеся заболевания, их основные клинические проявления.
Синдром Гольденхара (гемифациальная микросомия, окулоаурикуловертебральная дисплазия) Omim 164210.
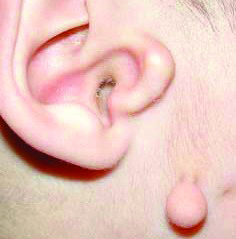
Частота развития заболевания — 1:3000 — 1:5000 новорожденных. Этиология заболевания остается до конца неизвестной. Большинство случаев — спорадические. Хотя выявлены семьи, у которых этот синдром передается по аутосомно-рецессивному и аутосомно-доминантному типу наследования. У нескольких детей, имеющих клиническую симптоматику, сходную с Синдромом Гольденхара, были выявлены хромосомные перестройки на 5, 8 и 21 хромосомах. В нескольких спорадических случаях синдрома Гольденхара были выявлены мутации в Гене GSC, который является кандидатным геном для гена HFM. У пяти пациентов — в гене TCOF1, мутации в котором приводят к развитию синдрома Тричера-Коллинза. Основные клинические проявления заболевания: Гипоплазия скуловой или нижниечелюстной области; Макростомия, асимметрия рта (вытянут в оду сторону); Гипоплазия мимических мышц; Микротия; Преаурикулярные выросты, ямки; Атрезия наружного слухового прохода; Кондуктивная тугоухость; Нейросенсорная тугоухость; Эпибульбарные дермоиды; Колобомы верхнего века; Микрофтальм; В 70% случаев поражение одностороннее; Гипоплазия позвонков; Клиновидные позвонки; В некоторых случаях — умственная отсталость, врожденные пороки сердца.
Гемифациальная микросомия с пороками конечностей (141400 Omim).
Тип наследования предположительно аутосомно-доминантный; Большинство случаев — спорадические; Врожденные пороки конечностей; Трехфаланговые пальцы; Удвоение фаланг; Расщелина мягкого и твердого неба; Гемифациальная микросомия; Преаурикулярные выросты; Кожные выросты в области нижней челюсти; Микротия; Атрезия наружного слухового прохода; Кондуктивная тугоухость; Данный синдром может рассматриваться как вариант Синдрома Гольденхара.
Бранхио-ото-ренальный синдром (Синдром Менльника-Фрейзера)(Omim 113650).
Тип наследования — аутосомно-доминантный; Заболевание обусловлено мутациями в гене EYA1, расположенного на длинном плече хромосомы 8 (8q13.3); Частота заболевания 1:40000 новорожденных; Длинное лицо; У 10 % пациентов -парез лицевого нерва; Нейросенсорная тугоухость (20%); Кондуктивная тугоухость (30%); Смешанная тугоухость (50%); Микротия; Чашеобразные уши; Деформация мочки, наружного уха; Бранхиогенные кисты или свищи; Стеной наружного слухового прохода; Пороки развития среднего и внутреннего уха; Атрезия или сужение слезного канала; Дисплазия Мондини; Прогнатия; Высокое небо; Расщелины твердого и мягкого неба; Расщепление небного язычка; Жаберные щели, расположенные симметрично с обеих сторон — редко; Дисплазия почек; Аплазия почек; Поликистоз почек; Пороки собирательной системы почек; Врожденный вывих бедра; Незавершенный поворот кишечника.
Бранхио-отический синдром (Omim 602588).
Тип наследования — аутосомно-доминантный; Заболевание обусловлено мутациями в гене EYA1; Нейросенсорная тугоухость; Кондуктивная тугоухость; Смешанная тугоухость; Микротия; Чашеобразные уши; Деформация мочки, наружного уха; Бранхиогенные кисты или свищи; Стеной наружного слухового прохода; Пороки развития среднего и внутреннего уха; Данный синдром иногда рассматривают, как вариант синдрома Мельника -Фрезера, но большинство исследователей склоняются к тому, что это разные заболевания, вызванные аллельными мутациями гена EYA1.
Бранхио-окуло-фациальный синдром (Omim 113620).
Тип наследования — аутосомно-доминантный. Заболевание обусловлено мутациями в гене TFAP2A, расположенного на коротком плече 6 хромосомы — 6р24.3. Основные клинические проявления заболевания: Пренатальная задержка роста; Задержка роста; Бронхиогенные кисты или свищи; Атрофия или очаговая аплазия кожи; Гемангиомы на коже; Атрезия слезного протока; Колобомы; Микрофтальм; Анофтальмия; Гамартома сетчатки; Дермоидные кисты глазницы; Эпителиальные кисты радужной оболочки; Катаркта; Миопия; Монголоидный разрез глаз; Телекант; Миопия; Низкопосаженные, ротированнын ушные раковины; Гипоплазия верхней части завитка; Микротия; Преаурикулярные выросты; Кондуктивная тугоухость; Аномалии среднего уха; Свищи над ушами; Широкое, запавшее переносье; Псвевдо-расщелины, расщелины губ, неба; Аномалии зубов; Микрогнатия; Гипоплазия скуловых костей; Агенезия почек; Дисплазия почек; Клинодактилия, полидактилия, грубое нарушение дерматоглифики; Гипоплазия ногтей; Эктопия тимуса; Ранняя седина; Умственная отсталость легкой степни; Гнусавый голос.
Синдром микротии нарушения слуха и расщелины неба (Omim 612290).
Тип наследования: аутосомно-доминантный, аутосомно-рецессивный. Заболевание обусловлено мутациями в гене HOXA2, расположенном на коротком плече 7 хромосомы — 7р15.2. Основные клинические проявления заболевания: Микротия; Сужение наружного слухового прохода (выраженное — при аутосомно-рецессивных формах); Деформация слуховых косточек; Заполнение барабанной полости костной тканью; Сращение слуховых косточек (при аутосомно-рецессивных формах); Кондуктивная тугоухость; Полная потеря слуха (при аутосомно-рецессивных формах); Расщелина твердого и мягкого неба.
Синдром Таунза-Брукса (Omim 107408).
Тип наследования — аутосомно-доминантный. Заболевание обусловлено мутациями в гене SALL1, который расположен на длинном плече 16 хромосомы 16q12.1. Основные клинические проявления заболевания: Нейросенсорная тугоухость; Смешенная тугоухость; Маленькие, чашеобразные уши со свисающим завитком; Преаурикулярные выросты; Трехфаланговый, раздвоеннный широкий 1 палец; Преаксиальная полидактилия; Псевдоэпифиз 2-ой пястной кост; Сращение костей кистей и стоп; Гипоплазия или аплазия 3 пальцев; Клинодактилия 5 пальцев; Эктопия или атрезия заднего прохода; Врожденные пороки влагалища и прямой кишки; Агенезия или гипоплазия почек, поликистоз почек; Пузырно-мочеточниковый рефлюкс; Расщепление мошонки; Гипоспадия; Иногда — умственная отсталость.
Синдром Фрейзера (синдром Фрайзера) (Omim 219000).
Тип наследования — аутосомно-рецессивный. Заболевание обусловлено мутациями в генах FRAS1, расположенного на длинном плече 4 хромосомы — 4q21.21, GRIP1, расположенного на длинном плече 12 хромосомы — 12q14.3, и гена FREM2, расположенного на длинном плече 13 хромосомы — 13q13.3. Основные клинические проявления заболевания: Двусторонний криптофтальм; Недоразвитие глазного яблока; Низкая граница роста волос на лбу на стороне поражения; Западение лобной кости на стороне поражения; Широкий нос; Запавшее переносье; Недоразвитие крыльев носа; Чашеобразные ушные раковины; Микроотия; Атрезия слухового прохода; Пороки среднего уха; Кондуктивная тугоухость; Расщелина твердого и мягкого неба; Стеноз трахеи; Атрезия трахеи; Кожная синдактилия на кистях и стопах; Крипоизм; Гипоспадия; Атрезия влагалища; Агенезия почек; Микроцефалия; Менингоцеле; Энцефалоцеле; Более 20% детей погибают в первый год жизни.
Синдром Тричера-Коллинза (Синдром Франческетти, челюстно-лицевой дизостоз) (Omim 154500).
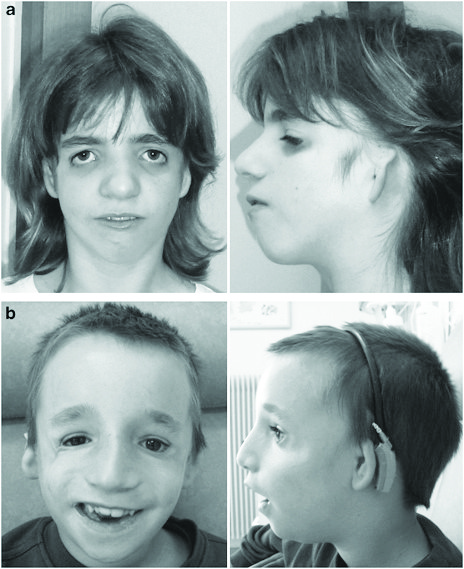
Тип наследования — аутосомно-доминантный. Заболевание обусловлено мутациями в гене TCQF1, расположенного на длинном плече 5 хромосомы — 5q31.3-q32. В настоящее время три типа данного заболевания. Основные клинические проявления заболевания: Антимонголоидный разрез глаз; Гипоплазия скуловых костей; Микрогнатия; Колобома нижнего века; Отсутствие ресниц на нижнем веке; Аномалии ушных раковин; ВПР наружного слухового прохода; Кондуктивная тугоухость; Расщелина неба; Нарушения глотания; Низкая граница роста волос на висках; Гипоплазия средней трети лица; Врожденные пороки сердца; Крипторхизм.
Синдром Пендреда (Omim 274600).
Тип наследования — аутосомно-рецессивный. Заболевание обусловлено мутациями в гене SLC26A4, расположенном на длинном плече 7 хромосомы, 7 q22.3. Основные клинические проявления заболевания: Нейросенсорная тугоухость; Аномалии развития структур костного лабиринта внутреннего уха; Неполное разделение завитка улитки; Дисплазии лабиринта улитки; Широкий водопровод преддверия; В подростковом возрасте — увеличение размеров щитовидной железы; Гипотиреоз; Карцинома щитовидной железы.
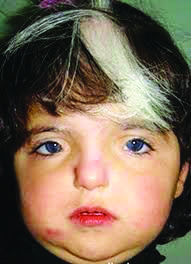
Тип наследования для I, II и III типов — аутосомно-доминантный, для четвертого — аутосомно-рецессивный. Основные клинические проявления заболевания: Короткие глазные щели; Телекант; Нос прямой; Переносье широкое, высокое; Гипоплазированные крылья носа; Густые брови; Синофриз; Очаги депигментации на коже; Бесцветные ресницы и брови; На глазном дне — картина, характерная для альбинизма; Седая прядь волос на лбу; Аплазия заднего полукружного канала; Нейросенсорная тугоухость; Выступающая нижняя челюсть; Высокое стояние лопаток; Добавочные ребра; Spina bifida; Врожденные пороки сердца.
В заключение хотелось бы сказать, что при подозрении у ребенка на наследственную патологию, сопровождающуюся различными формами нарушения слуха, ребенок должен получить консультацию врача-генетика, и при необходимости ему будет проведено молекулярно-генетическое обследование, исследование кариотипа для установления точного диагноза наследственного заболевания, уточнения прогноза развития заболевания и возможности медико-генетического консультирования семьи.
Читайте также: