
Секвенирование по сэнгеру кратко
Обновлено: 04.07.2024
Процесс, используемый для секвенирования ДНК, известен как метод обрыва цепи или метод Сэнгера. Он основан на модифицированной форме полимеразной цепной реакции.
Метод обрыва цепи (секвенирование по Сэнгеру)
Наряду с нуклеотидами и полимеразой, используемой в стандартном процессе ПЦР, подготовленная для реакции обрыва цепи среда содержит кроме четырех обычных нуклеотидов ДНК также и варианты каждого из них, так называемые дидезоксинуклеотиды.
Эти дидезоксинуклеотиды похожи на правильные нуклеотиды ДНК, но не содержат 3'-гидроксильной группы.
После того как дидезоксинуклеотид присоединен к растущей цепочке ДНК, ДНК-полимераза не может добавлять никаких других нуклеотидов. Таким образом, процесс репликации прекращается и полученный фрагмент ДНК прерывается.
Репликационная среда содержит только небольшое количество дидезоксинуклеотидных вариантов каждого из четырех нуклеотидов ДНК.
Во время прохождения полимеразной цепной реакции существует высокая вероятность того, что фермент полимеразы добавит немодифицированный нуклеотид к растущей цепи и что процесс репликации будет продолжаться. Но иногда полимераза связывает дидезоксинуклеотид с цепью и реакция прекращается.
В суспензии, содержащей миллиарды фрагментов ДНК, конечным результатом является серия фрагментов, заканчивающихся одним из четырех дидезоксинуклеотидов.
Все вместе эти фрагменты представляют собой все возможные расположения нуклеотидов A, C, G и T на синтезируемой нити.
Каждый из четырех вариантов дидезоксинуклеотида возможно пометить разными маркерами (например, красителем, который испускает свет с определенной длиной волны в ультрафиолетовом свете) для того, чтобы сделать различные нуклеотиды легко идентифицируемыми.
Поскольку фрагменты разделяются по длине и массе во время гель-электрофореза, маркеры указывают каким нуклеотидом заканчивается каждый фрагмент. Затем гель можно считывать снизу вверх для идентификации нуклеотидной последовательности.
Этот этап обычно включает в себя использование автоматического секвенатора ДНК, который ускоряет процесс считывания.
Метод секвенирования по Сэнгеру может быть использован для того, чтобы определить за одну реакцию последовательность ДНК образца, содержащего до одной тысячи пар оснований.
Этапы секвенирования больших геномов
Секвенирование большого генома включает в себя следующие три основных этапа:
1. Картирование генома.
Весь геном сначала случайно разбивается на более мелкие последовательности ДНК - приблизительно от 100 000 до 300 000 пар оснований.
Эти участки затем клонируют в бактериальном векторе, называемом бактериальной искусственной хромосомой или ВАС.
Повторяя этот цикл несколько раз, исследователи получают серию перекрывающихся ВАС.
Затем эти ВАС пропускают через гель-электрофорез, чтобы определить их индивидуальные фингерпринты ДНК. Изучив структуру этих паттернов, исследователи могут определить исходный порядок ВАС в геноме.
2. Секвенирование ДНК.
После того как исходный порядок ВАС был картирован, каждый ВАС разбивается рестрикционными эндонуклеазами на гораздо более мелкие фрагменты, которые могут быть секвенированы с использованием реакции обрыва цепи.
Этот этап секвенирования иногда называют BAC-BAC секвенирование.
3. Анализ результатов.
Паттерн перекрывающихся последовательностей ДНК используется для определения порядка фрагментов в каждом ВАС.
В этой процедуре используется несколько различных компьютерных программ, которые могут анализировать последовательности ДНК.
Секвенирование целого генома (метод дробовика, шотган-секвенирование, shotgun sequencing)

Этот метод полностью пропускает стадию картирования генома.
Вместо этого весь геном разбивается на случайные фрагменты сначала около 2000, а затем около 1000 пар оснований.
(Наличие фрагментов различной длины помогает сделать сборку нуклеотидных последовательностей более точной.)
Эти фрагменты, которые насчитывают миллионы, затем секвенируются и анализируются, после чего определяются нуклеотидные последовательности, соответствующие каждой из хромосомам.
Все это делается с помощью мощных компьютеров и сложных программ.
Секвенирование следующего поколения (высокопроизводительное секвенирование) (Next-generation sequencing, high-throughput sequencing)
Секвенирование следующего поколения (NGS) - это термин, используемый для описания ряда различных современных технологий секвенирования которые включают:
- Illumina (Solexa) sequencing
- Roche 454 sequencing
- Ion torrent: Proton / PGM sequencing
- SOLiD sequencing

Платформы NGS выполняют массовое параллельное секвенирование, в ходе которого миллионы фрагментов ДНК из одного образца секвенируются одновременно.
Технология массового параллельного секвенирования обеспечивает высокопроизводительное секвенирование, что позволяет секвенировать весь геном менее чем за один день.
В последнее десятилетие были разработаны несколько платформ NGS, которые обеспечивают недорогое и высокопроизводительное секвенирование.
Проект генома человека
Полный сиквенс человеческого генома был впервые опубликован в феврале 2001 года. Этот сиквенс был первым геномом млекопитающих, у которого была определена последовательность нуклеотидов в каждой хромосоме.
В результате проекта генома человека (HGP) была определена последовательность трех миллиардов пар оснований, которые составляют геном человека.
Среди непосредственных результатов проекта было открытие, что ДНК всех людей (Homo sapiens) более чем на 99,9% идентичны.
Другими словами, это означает, что все различия между людьми во всем мире являются результатом изменений менее чем одного нуклеотида на каждую 1000 в геноме каждого человека.
Цели генетических исследований

История проекта

Что же такое секвенирование?
Секвенирование – это заключительная ступень анализа генома человека. Перед этим следуют такие стадии: материал отбирается и клонируется. А также происходит предварительное тестирование участка ДНК более простыми методами. Секвенирование – это заключительное определение нуклеотидного ряда молекулы ДНК. Всего существует два способа этого генетического исследования: во-первых, используется метод Максама-Гилберта. Он базируется на способах расщепления молекулы ДНК по одному основанию. Есть более простой способ, который в практике используется чаще. Это так называемый дидезокси-метод, или секвенирование по Сэнгеру.
Стадии расшифровки генома по Сэнгеру
Последний метод включает в себя следующие стадии исследования:
- изучаемый фрагмент молекулы ДНК гибридизируется с праймером;
- происходит ферментативный синтез молекулы;
- на следующей стадии материал подвергается электрофорезу;
- и, наконец, полученные результаты анализируются исследователями-генетиками на радиоавтографе.
Большая часть этих приборов способна распознавать до 300 полос в молекуле ДНК.
Процесс дешифровки ДНК по Сэнгеру
Первоначальным способом секвенирования, который научные работники смогли использовать с целью обработки геномов, стало секвенирование по Сэнгеру (Sanger sequencing). Суть его состоит в следующем: ДНК клонируется, а затем полученная смесь делится на четыре части. Каждая из них помещается в специальный раствор, в котором присутствуют следующие вещества:
- молекулы ДНК-полимеразы;
- праймеры, которые способствуют процессу репликации;
- смесь из четырех копий нуклеотидов;
- отдельные вариации оного из нуклеотидов.
Таким образом, расшифровка генома по Сэнгеру практически идентична с процессом клонирования ДНК человека. Отличаются эти процессы только тем, что в нуклеотиды подмешиваются ложные компоненты.
Процесс расшифровки сегодня

Станет ли это доступным?
Сейчас ведущими странами, которые производят секвенирование ДНК, являются Китай и Соединенные Штаты Америки. Они находятся в ситуации конкуренции – победит та страна, которая первой сможет расшифровать человеческий геном, потратив на это менее сотни долларов. Американская исследовательская компания под названием Ilumina – вот главный противник Китая в этом вопросе. Китайскими учеными еще в 2010 году было закуплено первоклассное оборудование, которое позволяло им занимать все эти годы лидирующие позиции на рынке по услугам секвенирования генома. Но, во-первых, эти приборы стали неизбежно устаревать, а, во-вторых, услуга по расшифровке ДНК становится все более и более дешевой. Сейчас Китай не хочет тратить и без того недостающие финансовые средства на оборудование из-за рубежа – ученые решили разработать собственную аппаратуру. Кто победит в схватке за лидерство – покажет будущее. Но уже сегодня расшифровка генома в отдельных случаях стоит не более 600 долларов.
Действительно ли расшифровка генов полезна?
Впрочем, не все так просто в этом вопросе. Некоторые исследователи настаивают на том, что секвенирование генов приносит не столько пользу, сколько вред. Может ли этот процесс действительно что-то дать отдельному индивиду или быть вкладом в науку? Почему часть специалистов относится к расшифровке ДНК более чем критически?
Дело в том, что методы секвенирования, с их точки зрения, являются не более чем суперсовременным способом гадания на кофейной гуще. Ответственность за получение информации о своем геноме человек несет сам. Разные мотивы толкают людей на ответы на вопросы о своем ДНК. Кто-то стремится таким образом лучше узнать себя. Кто-то пытается себя обезопасить от заболеваний – и делает все, чтобы перестраховаться. Однако такой подход может быть крайне негативным.
Например, люди, которые узнают, что имеют риск заболеть онкологией, в дальнейшем начинают страдать от серьезных депрессий и без конца бегать по врачам. И это при том, что даже полное секвенирование дает результаты, которые прогнозируют развитие страшных заболеваний с риском 50%. Однако среди клиентов этих услуг все больше распространяется волна истерии. Все это привело к тому, что некоторые власти, например, в Соединенных Штатах, серьезно взялись за ограничение деятельности медицинских компаний.
Генетический паспорт – да или нет?
Возможные ошибки
![]()
Геномика: постановка задачи и методы секвенирования

Сергей Николенко, кандидат физико-математических наук, старший научный сотрудник лаборатории вычислительной биологии Санкт-Петербургского Академического Университета в серии статей говорит о некоторых задачах биоинформатики, связанных со сборкой и анализом геномов, делая акцент на математической, комбинаторной постановке задачи. В данном, вводном, тексте речь идет о том, как выглядят входные данные для сборки геномов и как их получают.
Как выглядит молекула ДНК?

Рисунок из Википедии
Что такое секвенирование?
Клонирование происходит либо просто выращиванием клеток в чашке Петри, либо (в случаях, когда это было бы слишком медленно или по каким-то причинам не получилось бы) при помощи так называемой полимеразной цепной реакции. В кратком и неточном изложении работает она примерно так: сначала ДНК денатурируют, т.е. разрушают водородные связи, получая отдельные нити. Затем к ДНК присоединяют так называемые праймеры; это короткие участки ДНК, к которым может присоединиться ДНК-полимераза – соединение, которое, собственно, и занимается копированием (репликацией) нити ДНК.

Рисунок из Википедии
На следующем этапе полимераза копирует ДНК, после чего процесс можно повторять: после новой денатурации отдельных нитей будет уже вдвое больше, на третьем цикле – вчетверо, и так далее.
Секвенирование по Сэнгеру
Первым методом секвенирования, который учёные сумели применить для обработки целых геномов (в том числе генома человека), стало секвенирование по Сэнгеру (Sanger sequencing). Смысл таков: участок ДНК клонируется, после чего полученная смесь делится на четыре части. Каждая часть помещается в активную среду, где присутствуют:
ATGCAGAACAGACGATCAGCGACACTTTA (образец)
AT
ATGCAGAACAGACGAT
ATGCAGAACAGACGATCAGCGACACT
ATGCAGAACAGACGATCAGCGACACTT
ATGCAGAACAGACGATCAGCGACACTTT
Очевидно, что эта последовательность начинается с А (т.к. самый лёгкий префикс, из одной буквы, заканчивается на A); дальше идёт C, дальше опять A, и так далее. В результате можно прочесть исходный участок: ATGCAGAACA.

Рисунок из Википедии
Видно, что (в идеальном случае) можно просто прочесть последовательность нуклеотидов от самого лёгкого префикса (т.е. префикса из одной буквы) к самому тяжёлому.
Результаты и ошибки сэнгеровского секвенирования
На выходе из сэнгеровского секвенатора получаются короткие участки ДНК, так называемые риды (reads). Для биоинформатики принципиальны две вещи: во-первых, какой длины получаются риды, во-вторых, какие в них могут быть ошибки и как часто (разумеется, на свете нет ничего идеального).
Сэнгеровские риды по этим критериям очень хороши: получаются риды длиной около тысячи нуклеотидов, причём качество начинает заметно падать только после 700-800 нуклеотидов. Сам процесс секвенирования по Сэнгеру, с которым мы познакомились в предыдущем разделе, предопределяет и эффект падения качества (труднее отличить молекулу массой 700 от молекулы массой 701, чем массу 5 от массы 6), и другой неприятный эффект – если в геноме встречается длинная последовательность из одной и той же буквы (…AAAAAAAA…), трудно бывает точно определить, какой она длины – все промежуточные массы попадут в одну и ту же пробирку, некоторые из них могут не встретиться, некоторые слиться друг с другом и т.д. Но всё же сэнгеровское секвенирование даёт отличные результаты с достаточно длинными ридами, которые потом относительно легко собирать. О том, как это делается, мы будем говорить в последующих текстах.
Именно при помощи сэнгеровского секвенирования был впервые расшифрован геном человека. Секвенирование по Сэнгеру применяется и сегодня, но его всё активнее вытесняют другие методы, и применяется оно всё реже. Кому же и почему оно уступило свои позиции?
Секвенаторы второго поколения: Illumina
Современные секвенаторы – это так называемые секвенаторы второго поколения (SGS, second generation sequencing). В них участки ДНК по-прежнему многократно клонируются, но процесс чтения устроен не так, как у Сэнгера. Существует много разных методов, отличающихся довольно существенно, поэтому мы рассмотрим только один из них, один из самых популярных на сегодня – секвенирование по методу Solexa (ныне Illumina; в смене названия не нужно искать глубокий смысл, просто одна компания купила другую).
Процесс секвенирования Illumina проиллюстрирован на рисунке; кроме того, можно посмотреть один из нескольких существующих видеороликов с анимацией этого процесса – в данном случае, действительно, лучше один раз увидеть, чем сто раз прочесть текст. Однако краткие комментарии тоже пригодятся; вот как происходит процесс секвенирования по методу Illumina.
В результате на каждом цикле мы прочитываем одновременно очень большое число нуклеотидов из разных последовательностей. Но за это приходится платить тем, что участки ДНК, которые мы можем прочесть, оказываются гораздо короче, чем в случае секвенирования по Сэнгеру – риды Illumina обычно получаются длиной около 100 нуклеотидов.
Парные риды и постановка задачи
Итак, теперь мы можем формально поставить задачу сборки геномов. Она звучит так: по большому числу подстрок небольшой длины восстановить исходную длинную строку в алфавите из букв A, C, G, T. В случае секвенирования по методу Illumina – по большому числу пар коротких подстрок, разделённых в исходной строке приблизительно известным расстоянием. Поставив эту задачу, мы можем забыть про биологию, химию и медицину – перед нами чисто алгоритмическая задача. Однако, прежде чем перейти к математике, сделаем ещё несколько замечаний.
Ошибки и показатели качества в секвенаторах второго поколения
Как мы уже знаем, секвенирование всегда содержит ошибки. В секвенаторах Illumina и аналогичных ошибки, как правило, происходят на фазе, когда нужно распознать помеченные нуклеотиды, т.е. понять, каким цветом и с какой силой светятся кластеры из многократно клонированных участков ДНК. На рисунке – типичный пример такой фотографии, порождённой секвенатором Illumina.

Рисунок с сайта medicine.yale.edu
Проблема здесь заключается в том, что из-за неидеальности остальных этапов процесса кластеры никогда не светятся только одним цветом; это всегда смесь всех четырёх цветов с той или иной интенсивностью. Нужно выделить наиболее интенсивную компоненту и оценить, насколько вероятна ошибка в этой букве; эта задача называется base calling (распознавание нуклеотидов). Base calling – это целая наука, в подробности которой мы сейчас вдаваться не будем.
Для нас сейчас важно, что в результате каждому нуклеотиду каждого рида секвенатор ставит в соответствие вероятность того, что этот нуклеотид был распознан правильно. Эти вероятности тоже можно использовать при сборке, и секвенаторы выдают их вместе с собственно ридами.
В итоге типичный рид в так называемом fastq-формате, стандартном для секвенаторов второго поколения, выглядит примерно так:
Первая и третья строки содержат имя рида; вторая строка – сама последовательность нуклеотидов. Обратим внимание, что среди букв A, C, G, T встречаются и буквы N – это значит, что секвенатор не смог однозначно определить, какой здесь был нуклеотид, и сдался. А четвёртая строка кодирует, в логарифмическом масштабе, вероятности того, что тот или иной нуклеотид распознан правильно; например, H здесь соответствует вероятности ошибки около одной десятитысячной. Как правило, качество ухудшается к концу рида; в нашем примере, как видите, хвост рида и вовсе не удалось сколь-нибудь надёжно прочитать.
Другие методы секвенирования
Хотя мы подробнее всего рассмотрели секвенатор Illumina (Solexa), на самом деле на этом методе свет клином не сошёлся. Есть и другие секвенаторы второго поколения, с другими свойствами.
Пиросеквенирование (pyrosequencing) основано на хемилюминесцентных сигналах, которые подают специально модифицированные нуклеотиды, когда соединяются с комплементарным нуклеотидом в прочитываемой нити ДНК; на этом принципе работает, например, секвенатор 454 от 454 Life Sciences.
Недавно появившийся метод ионного полупроводникового секвенирования (на нём основан секвенатор IonTorrent) вместо всего этого просто детектирует соединения (ионы), которые выделяются при присоединении нового нуклеотида к нити ДНК. Это позволяет радикально сократить время и стоимость получаемых ридов, хотя процент ошибок становится больше, и больше становится ошибок в фрагментах из повторяющейся одной буквы.
Человеческая мысль не стоит на месте: методы секвенирования постоянно улучшаются. Однако практически все современные методы выдают относительно короткие риды, от 100 до 400 нуклеотидов; в этом цикле мы будем в основном говорить о том, как собирать именно короткие риды.
Sanger или Illumina?
Человеческий геном был впервые собран на сэнгеровских секвенаторах, причём алгоритмическая сторона того проекта была проработана гораздо меньше, чем сейчас, десять лет спустя. Алгоритмы, которыми собирали первый человеческий геном, значительно проще тех, о которых мы будем говорить в дальнейшем. Однако первый геном всё-таки собрали; может быть, весь алгоритмический прогресс – это никому не нужный миф, и вполне хватило бы старых программ?
На таком уровне становится важной и цена алгоритмической стороны вопроса. Чтобы сборка геномов не занимала дольше и не стоила дороже, чем само их секвенирование, нужно разработать очень быстрые алгоритмы для решения задачи сборки. Об этом пойдет речь в следующей статье.
Секвенирование - представляет собой определение нуклеотидной последовательности фрагмента ДНК путем получения серии комплементарных молекул ДНК, различающихся по длине на одно основание. Является последним этапом молекулярного анализа предварительно отобранного, клонированного и протестированного более простыми методами фрагмента ДНК.
Существует два основных метода секвенирования; метод Максама-Гилберта (основан на химическом расщеплении ДНК по одному основанию) и метод Сэнгера (дидезокси-метод). Метод Сэнгера более надежен и прост в исполнении, и на практике его используют чаше.
Метод Сэнгера или дидезоксисеквенирование основан на синтезе изучаемой цепи ДНК in vitro с остановкой синтеза на заданном основании путем присоединения дидезоксинуклеотида. Дидезоксинуклеотид лишен гидроксильных групп при атомах сахарного кольца не только в 2'-, но и в 3'-положении, что делает его неспособным формировать фосфодиэфирную связь со следующим нуклеотидом.
1) секвенирующий праймер (искусственно синтезированная олигонуклеотидная последовательность, комплементарная определенному участку исходной молекулы ДНК),
2) набором из четырех дезоксинуклеотидов dATP, dCTP, dGTP и dTTP, один из которых изотопно меченный
3) один из четырех дидезоксинуклеотидов (ddATP, ddCTP, ddGTP и ddTTP)
Сам метод включает следующие этапы:
1) гибридизация изучаемого фрагмента ДНК с праймером
3) денатурация полученных продуктов формамидом (в результате образуются уникальные различающиеся по длине олигонуклеотидные последовательности, содержащие праймер)
5) анализ результатов на радиоавтографе. На большинстве радиоавтографов можно четко различить 250—350 полос, т.е. прочитать последовательность в 250-350 п.н. Нуклеотидная последовательность на радиоавтографе считывается снизу вверх.
Таким образом, по размеру синтезированных фрагментов может быть определена локализация дидезоксинуклеотидов и порядок соответствующих им нуклеотидов в исходной молекуле ДНК.
Для прочитывания более длинных последовательностей существует ряд методов, представляющих собой разные модификации метода Сэнгера. Эти методы основаны на предварительном клонировании ДНК в векторах, сконструированных на основе фага М13 E.coli для получения протяженных одноцепочечных участков ДНК, которые могут быть непосредственно секвенированы без денатурации и праймирования. Особенность фага М13 E.coli состоит в возможности его существования в двух формах: двухцепочечной репликативной, функционирующей как плазмида, и одноцепочечной фаговой, использующейся в качестве матрицы для секвенирования. Выделив после клонирования одноцепочечные фаговые ДНК со вставкой (размер около 500 п.н.), праймер гибридизуют с последовательностью вблизи вставки, и проводят дидезоксисеквенирование.
Для секвенирования крупных фрагментов ДНК (около 2000 п,н.) используют более сложные (комбинированные) подходы. Один из них заключается в предварительном клонировании данного фрагмента в плазмидном векторе и построении его подробной рестрикционной карты, идентификации перекрывающихся рестрикционных фрагментов длиной 100-500 п.н. Далее, субклонировав каждый из этих фрагментов в ДНК M13 E.coli, их секвенируют и определяют последовательность исходного участка ДНК. Так как субклонированные фрагменты могут быть встроены в противоположных направлениях, то праймер в одном случае будет инициировать синтез первой цепи, а в другом - комплементарной ей, а значит возможно одновременное секвенирование обеих цепей.
1) отжиг плазмидной ДНК, содержащей вставку, с праймером, комплементарным последовательности одной из цепей векторной ДНК;

2) дидезоксисеквенирование и идентификация первых 250-350 п.н. вставки;
3) синтез второго праймера, комплементарного сегменту вставки, который отстоит от места связывания первого праймера примерно на 300 п.н., и секвенирование следующих 250-350 п.н., и так далее, пока не секвенируют весь фрагмент.
Обязательное условие при секвенировании очень длинных фрагментов иметь праймер длиной не менее 24 нуклеотидов. Это необходимо для того, чтобы избежать спаривания праймера внутри вставки более одного раза.
Однако существуют еще более быстрые методы автоматического секвенирования. Один из них - секвенирование путем гибридизации исследуемой последовательности ДНК с набором олигонуклеотидов (олигонуклеотидной матрицей), включающим все возможные варианты перестановок из 4 стандартных нуклеотидов (А, G, С, Т) определенной длины. Наиболее удобными считаются наборы матриц (чипы) из октануклеотидов., при этом количество возможных вариантов нуклеотидов составляет 65536. Секвенируемый фрагмент ДНК, меченный радиоактивным фосфором, гибридизуется только с комплементарными его участкам октануклеотидами.
В результате определяется спектр октануклеотидов, составляющих исследуемый фрагмент ДНК. Локализация октамеров в изучаемом фрагменте ДНК устанавливается при помощи специальной компьютерной программы.
Секвенирование - представляет собой определение нуклеотидной последовательности фрагмента ДНК путем получения серии комплементарных молекул ДНК, различающихся по длине на одно основание. Является последним этапом молекулярного анализа предварительно отобранного, клонированного и протестированного более простыми методами фрагмента ДНК.
Существует два основных метода секвенирования; метод Максама-Гилберта (основан на химическом расщеплении ДНК по одному основанию) и метод Сэнгера (дидезокси-метод). Метод Сэнгера более надежен и прост в исполнении, и на практике его используют чаше.
Метод Сэнгера или дидезоксисеквенирование основан на синтезе изучаемой цепи ДНК in vitro с остановкой синтеза на заданном основании путем присоединения дидезоксинуклеотида. Дидезоксинуклеотид лишен гидроксильных групп при атомах сахарного кольца не только в 2'-, но и в 3'-положении, что делает его неспособным формировать фосфодиэфирную связь со следующим нуклеотидом.
1) секвенирующий праймер (искусственно синтезированная олигонуклеотидная последовательность, комплементарная определенному участку исходной молекулы ДНК),
2) набором из четырех дезоксинуклеотидов dATP, dCTP, dGTP и dTTP, один из которых изотопно меченный
3) один из четырех дидезоксинуклеотидов (ddATP, ddCTP, ddGTP и ddTTP)
Сам метод включает следующие этапы:
1) гибридизация изучаемого фрагмента ДНК с праймером
3) денатурация полученных продуктов формамидом (в результате образуются уникальные различающиеся по длине олигонуклеотидные последовательности, содержащие праймер)
5) анализ результатов на радиоавтографе. На большинстве радиоавтографов можно четко различить 250—350 полос, т.е. прочитать последовательность в 250-350 п.н. Нуклеотидная последовательность на радиоавтографе считывается снизу вверх.
Таким образом, по размеру синтезированных фрагментов может быть определена локализация дидезоксинуклеотидов и порядок соответствующих им нуклеотидов в исходной молекуле ДНК.
Для прочитывания более длинных последовательностей существует ряд методов, представляющих собой разные модификации метода Сэнгера. Эти методы основаны на предварительном клонировании ДНК в векторах, сконструированных на основе фага М13 E.coli для получения протяженных одноцепочечных участков ДНК, которые могут быть непосредственно секвенированы без денатурации и праймирования. Особенность фага М13 E.coli состоит в возможности его существования в двух формах: двухцепочечной репликативной, функционирующей как плазмида, и одноцепочечной фаговой, использующейся в качестве матрицы для секвенирования. Выделив после клонирования одноцепочечные фаговые ДНК со вставкой (размер около 500 п.н.), праймер гибридизуют с последовательностью вблизи вставки, и проводят дидезоксисеквенирование.
Для секвенирования крупных фрагментов ДНК (около 2000 п,н.) используют более сложные (комбинированные) подходы. Один из них заключается в предварительном клонировании данного фрагмента в плазмидном векторе и построении его подробной рестрикционной карты, идентификации перекрывающихся рестрикционных фрагментов длиной 100-500 п.н. Далее, субклонировав каждый из этих фрагментов в ДНК M13 E.coli, их секвенируют и определяют последовательность исходного участка ДНК. Так как субклонированные фрагменты могут быть встроены в противоположных направлениях, то праймер в одном случае будет инициировать синтез первой цепи, а в другом - комплементарной ей, а значит возможно одновременное секвенирование обеих цепей.
1) отжиг плазмидной ДНК, содержащей вставку, с праймером, комплементарным последовательности одной из цепей векторной ДНК;
2) дидезоксисеквенирование и идентификация первых 250-350 п.н. вставки;
3) синтез второго праймера, комплементарного сегменту вставки, который отстоит от места связывания первого праймера примерно на 300 п.н., и секвенирование следующих 250-350 п.н., и так далее, пока не секвенируют весь фрагмент.
Обязательное условие при секвенировании очень длинных фрагментов иметь праймер длиной не менее 24 нуклеотидов. Это необходимо для того, чтобы избежать спаривания праймера внутри вставки более одного раза.
Однако существуют еще более быстрые методы автоматического секвенирования. Один из них - секвенирование путем гибридизации исследуемой последовательности ДНК с набором олигонуклеотидов (олигонуклеотидной матрицей), включающим все возможные варианты перестановок из 4 стандартных нуклеотидов (А, G, С, Т) определенной длины. Наиболее удобными считаются наборы матриц (чипы) из октануклеотидов., при этом количество возможных вариантов нуклеотидов составляет 65536. Секвенируемый фрагмент ДНК, меченный радиоактивным фосфором, гибридизуется только с комплементарными его участкам октануклеотидами.
В результате определяется спектр октануклеотидов, составляющих исследуемый фрагмент ДНК. Локализация октамеров в изучаемом фрагменте ДНК устанавливается при помощи специальной компьютерной программы.

Предпосылками для формирования генетики как самостоятельной научной области послужило открытие законов Менделя. В дальнейшем в XX веке было сделано четыре открытия, положивших начало развитию генетики [1]:
• установлены клеточные основы наследственности — хромосомы;
• определена молекулярная основа наследственности — двойная спираль ДНК;
• открыта информационная основа наследственности, а также биологический механизм, с помощью которого клетки считывают информацию, содержащуюся в генах;
• изобретены технологии клонирования и секвенирования рекомбинантных ДНК.
Последняя четверть прошлого века была отмечена неустанным стремлением расшифровать сначала гены, а затем и целые геномы [2].
Первая рабочая концепция секвенирования — метод Сэнгера, также известный как метод обрыва цепи, — была предложена в 1977 году. За это открытие Фредерик Сэнгер был удостоен Нобелевской премии по химии в 1980 году. Этот метод секвенирования применялся в течение 40 лет, а его усовершенствование и коммерциализация привели к широкому распространению секвенирования [2].
Описание метода Сэнгера
Секвенирование Сэнгера — метод, при котором используются олигонуклеотидные праймеры для поиска определенных областей ДНК. Этот процесс начинается с деспирализации двухцепочечной ДНК [5]. Одна цепочка ДНК является матрицей для синтеза комплементарной цепочки при помощи фермента ДНК-полимеразы. Реакцию с одной и той же цепочкой проводят в четырех разных пробирках, каждая из которых содержит [3]:
— праймер;
— четыре дезоксинуклеотида (дезоксиаденозинтрифосфат, дезоксигуанозинтрифосфат, дезоксицитидинтрифосфат и тимидинтрифосфат);
— небольшое количество (1 к 100) одного из радиоактивно меченных дезоксинуклеотидов (для визуализации продуктов реакции).
В каждой пробирке образуется набор фрагментов ДНК разной длины, заканчивающихся одним и тем же нуклеотидом. После завершения реакции содержимое пробирок разделяют электрофорезом в полиакриламидном геле в денатурирующих условиях и затем проводят авторадиографию гелей. Каждый дезоксинуклеотид отмечен флуоресцентным маркером: A — зеленый цвет, T — красный, G — черный и C — синий. Лазер в автомате, используемый для считывания последовательности, фиксирует интенсивность флуоресценции. Когда в последовательности встречается гетерозиготный вариант, локусы захватываются двумя флуоресцентными красителями одинаковой интенсивности. Если присутствует гомозиготный вариант, ожидаемый флуоресцентный цвет заменяется цветом комплементарного основания [5].
Секвенирование Сэнгера — это надежный метод для определения генных мутаций, который широко использовался в течение нескольких десятилетий. Метод Сэнгера является геноспецифичным, и с его помощью анализируют небольшое подмножество генов. Секвенирование Сэнгера позволяет идентифицировать мозаичные мутации. Но метод секвенирования Сэнгера не позволяет проводить точную количественную оценку, то есть нельзя сделать вывод о том, в каком количестве клеток есть мутация [5].
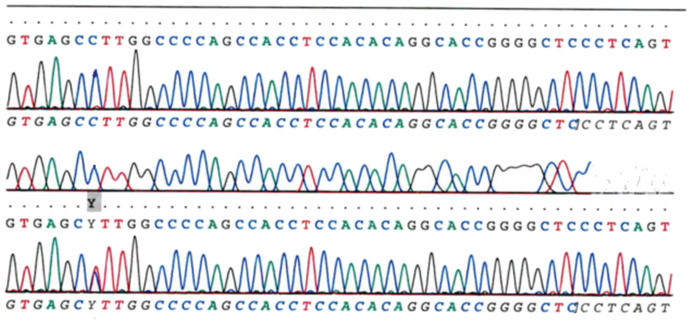
На электроферограмме отмечено изменение нуклеотида С на Т (мутация отмечена буквой Y) по сравнению с секвенированием контрольных образцов. Эта мутация является гетерозиготной, так как оба аллеля содержат разные нуклеотиды. Предоставлено доктором Людвином Мессианом, PhD, Университет Алабамы.
Метод дробовика
Метод дробовика используется для секвенирования длинных участков ДНК. Суть метода состоит в получении случайной массированной выборки клонированных фрагментов ДНК данного организма, на основе которых восстанавливается исходная последовательность ДНК [6].
Первые методы секвенирования способны восстанавливать небольшие последовательности ДНК (порядка 1000 нуклеотидов), следовательно, для секвенирования более длинных последовательностей требовалось разработать новый подход. При секвенировании методом дробовика ДНК случайным образом фрагментируется на мелкие участки с помощью сайт-неспецифичных нуклеаз. Затем фрагменты секвенируют любым доступным методом, например, методом секвенирования по Сэнгеру. Полученные перекрывающиеся случайные фрагменты ДНК собирают с помощью специального программного обеспечения в одну целую последовательность. Данный метод оставался фундаментальным методом секвенирования генома в течение 20 лет [2]. В 1981 году метод применен на практике — полное секвенирование генома вируса мозаики цветной капусты [7].
На практике трудности возникают из-за повторяющихся последовательностей. Например, можно легко секвенировать типичные бактериальные геномы (около 1,5 % повторов) или эухроматическую часть генома мухи (около 3 % повторов). Человеческий геном содержит более чем 50 % повторяющихся последовательностей. Такие особенности усложняют сборку правильной и законченной последовательности генома [2].
В дальнейшем этот подход совершенствовался: были улучшены механизмы фрагментации и клонирования ДНК. В 1990 году был предложен метод секвенирования парных прочтений. Результаты первого применения метода секвенирования парных концов на практике были опубликованы в 1990 году в работе, посвященной секвенированию человеческого гена гипоксантин-гуанинфосфорибозилтрансферазы [4].
При секвенировании парных прочтений ДНК разрезается на случайные фрагменты, которые затем группируются по весу и клонируются в векторах. Клоны секвенируют с обоих концов с использованием метода обрыва цепи, в результате которого образуются две коротких последовательности [4].
Иерархическое секвенирование методом дробовика
Данный метод — технически более сложный из-за необходимости обработки больших объемов данных. Это служит причиной тому, что метод иерархического секвенирования имеет более высокую стоимость [2].

Библиотека создается путем фрагментации секвенируемого генома и его клонирования в специальном векторе (в данном случае векторы BAC — искусственная бактериальная хромосома). Фрагменты геномной ДНК, представленные в библиотеке, затем объединяются в физическую карту, и отдельные клоны ВАС рандомно отбираются и секвенируются с помощью метода дробовика. Наконец, последовательности клонов собирают для восстановления последовательности генома.
Читайте также: