
Мышечная дистрофия дюшенна кратко и понятно
Обновлено: 14.05.2024
Что такое мышечная дистрофия Дюшенна?
Мышечная дистрофия Дюшенна — это серьезное заболевание, которое вызывает прогрессирующее ослабление мышц. Из-за нехватки дистрофина мышечные волокна ослабевают и уступают место волокнистой или жировой ткани, вызывая постепенное разрушение мышц.
Каковы причины мышечной дистрофии Дюшенна?
У каждого сына носителя есть риск 50%, что он будет подвержен этому заболеванию, а у каждой дочери – риск 50%, что она – носитель.
Консультация генетиков и обследование остальных членов семьи на риск быть носителями должны быть проведены как можно скорее, как только мальчику будет поставлен диагноз мышечной дистрофии Дюшенна. Вы можете обратиться к Вашему врачу-консультанту или семейному врачу.
Как распознают мышечную дистрофию Дюшенна?
У большинства мальчиков с мышечной дистрофией Дюшенна это заболевание не обнаруживают, пока не появятся соответствующие симптомы, за исключением тех случаев, когда в семье есть человек с таким заболеванием. Первые признаки мышечной дистрофией Дюшенна обычно появляются в возрасте от одного до трех лет и, как правило, связаны с нарушением функционирования мышц. Мальчики могут начать ходить позже, чем их сверстники, падать чаще или испытывать трудности с бегом, прыжками или подъемом. У них могут быть увеличены икроножные мышцы.
У некоторых мальчиков с мышечной дистрофией Дюшенна замедленная речь, что может стать первым признаком этого заболевания. При сдаче анализа крови обнаруживается высокое содержание белка под названием креатинкиназа (КК). КК, как правило, появляется в мышце, но при их повреждении, как в случае с мышечной дистрофией Дюшенна, он попадает в кровь. Ферменты печени (аминотрансферазы, ALT и AST) также часто находятся на высоком уровне, как следствие повреждения мышц, а не нарушений печени.
Мышечная дистрофия Дюшенна должна быть подтверждена генетическим анализом крови. Разные типы генетических анализов предоставляют специфические и подробные данные о мутации ДНК.
Генетическое подтверждение заболевание имеет решающее значение. Это позволяет семьям проводить пренатальную диагностику будущих беременностей, а также обследовать остальных членов семьи на случай, если они являются носителями мутации в гене дистрофина. Более того, генетическая диагностика поможет определить, может ли мальчик претендовать на участие в клиническом исследовании, которое начинается или запланировано.
Ваш врач также может посоветовать пункционную биопсию мышц, когда берут небольшой образец мышцы для анализа. Такие исследования могут предоставить информацию о том, какое количество белка дистрофина присутствует в клетках мышцы, а также в некоторых случаях помочь отличить мышечную дистрофию Дюшенна от слабой формы заболевания, известной как мышечная дистрофия Беккера. Однако, как правило, достаточно клинических признаков и генетического анализа, чтобы различить эти две формы, без необходимости мышечной биопсии.
Существует ли лечение или лекарство?
Пока не было изобретено лекарства для этого заболевания, но есть обнадеживающие исследования в этой области. Подход с привлечением множества различных специалистов, в том числе физиотерапевтов и специалистов по трудотерапии (occupational therapists — специалисты, помогающие людям с ограниченными возможностями освоиться в жизни) — это лучший способ контроля мышечной дистрофии Дюшенна.
Иметь доступ к специалистам из разных областей — жизненно важно для обеспечения человека с мышечной дистрофией Дюшенна всей необходимой комплексной поддержкой. Это означает, что у пациента должна быть возможность за одно посещение специализированного центра проконсультироваться у специалистов разных направлений: получить консультации по дыхательной системе, кардиосистеме и физиотерапии. Работая вместе, такие специалисты обеспечат более эффективный уход.
Важно регулярно проверяться у специалистов, чтобы принимать решения о новых методах лечения в самое подходящее время, и возможно, предвидеть и предотвращать нарушения. Рекомендуется посещать Вашего лечащего врача раз в шесть месяцев, а физиотерапевта – каждые три-четыре месяца.
Физиотерапевт даст рекомендации касательно вмешательств (таких как упражнения для развития гибкости), которые могут потребоваться. Важно позволить Вашему сыну быть как можно активнее, а Ваш физиотерапевт будет Вам помогать.
Стероиды (преднизон или дефлазакорт) часто назначаются при мышечной дистрофии Дюшенна, так как они замедляют на какое-то время процесс ослабевания мышц и нарушения подвижности и предотвращают или откладывают развитие осложнений. Однако есть множество возможных побочных эффектов, которые необходимо тщательно контролировать.
Продолжается интенсивное изучение возможного лечения мышечной дистрофии Дюшенна. Некоторые лекарства сейчас проходят клинические испытания.
Полезно иметь копию генетического отчета (с типом и расположением мутации в гене дистрофина, обнаруженной у Вашего ребенка). Это поможет определить, какое лекарство или испытание больше подходит для Вашего ребенка.
Реестр мышечной дистрофии Дюшенна предоставит свежую информацию о ходе клинических испытаний и может помочь определить, какие дети предположительно могут быть допущены к конкретным клиническим исследованиям. Ваши врачи расскажут Вам как зарегистрировать Вашего ребенка в этом реестре.
The North Star Adult Network, в которую входят эксперты-консультанты по нервно-мышечным заболеваниям, специалисты смежных профессий, сами пациенты, живущие с мышечной дистрофией Дюшенна, и организация Muscular Dystrophy UK, работает над улучшением стандартных методов ухода и поддержки для взрослых на территории Соединенного Королевства. Есть и педиатрическая версия — the North Star Project – которая работает над улучшением ухода за детьми с мышечной дистрофией Дюшенна.
Какой прогноз?
На ранних стадиях у мальчиков с мышечной дистрофией Дюшенна обнаруживаются признаки мышечной слабости, такие как: трудности при беге, прыжках, подъеме по лестнице или с пола. Они могут двигаться в манере Гауэра (необходимость поддерживать себя руками за бедра при подъеме с пола) и ходить вразвалку (на носочках и с изогнутой поясницей).
По мере развития мышечной слабости мальчикам становится сложнее ходить так же быстро или так же далеко, как другим детям, они могут начать падать. Они все еще могут подниматься по лестнице, но уже приставляя вторую ногу к первой.
Позже, когда ходьба будет даваться с большим трудом, мальчикам будет сложно подниматься по лестнице или вставать с пола.
Стероиды значительно меняют ход болезни. Они помогают восстановить мышечную силу на какой-то определенный период времени и откладывают тот момент, когда мальчикам может понадобиться инвалидное кресло.
Спрогнозировать, когда пациент начнет пользоваться инвалидным креслом, — сложно, поскольку каждый случай отличается один от другого.
С дальнейшим развитием мышечной слабости сложнее становится сохранять положение тела, могут возникнуть осложнения. Заболевание — серьезное и может сократить продолжительность жизни, но в настоящее время, с высокими стандартами медицинской помощи, большинство юношей с мышечной дистрофией достигают взрослой жизни.
У некоторых мальчиков с мышечной дистрофией Дюшенна могут появиться трудности с обучаемостью и/или поведением, что является результатом воздействия заболевания на мозг. Проблемы с обучением при мышечной дистрофии Дюшенна не прогрессируют, важно их сразу распознать и сообщить о них (например, в школу), чтобы обеспечить ребенка поддержкой, которая необходима ему для развития навыков и достижения его полноценной обучаемости.
Поддержка семьи необходима, также следует сообщать специалистам о некоторых особенностях поведения и обучения.
Мышечная дистрофия Дюшенна: причины, диагностика, лечение
Этиология и встречаемость мышечной дистрофии Дюшенна. Мышечная дистрофия Дюшенна (MIM №310200) — панэтническая Х-сцепленная прогрессирующая миопатия, вызванная мутациями в гене DMD. Встречаемость составляет приблизительно 1 на 3500 новорожденных мальчиков.
Патогенез мышечной дистрофии Дюшенна. Ген DMD кодирует дистрофии, внутриклеточный белок, экспрессирующийся преимущественно в гладких, скелетных и сердечной мышце, а также в некоторых нейронах мозга. В скелетной мускулатуре дистрофии составляет часть большого комплекса связанных с сарколеммой белков, обеспечивающих устойчивость сарколеммы.
Мутации в гене DMD, вызывающие миодистрофию Дюшенна, включают крупные делеции (60-65%), крупные дупликации (5-10%) и небольшие делеции, инсерции или замены нуклеотидов (25-30%). Самые крупные делеции происходят в одной из двух горячих точек. Нуклеотидные замены встречаются по всему гену, преимущественно в динуклеотидах CpG.
De novo мутации возникают со сравнимой частотой в ходе овогенеза и сперматогенеза; наиболее крупные делеции de novo возникают в овогенезе, тогда как большинство de novo нуклеотидных замен возникает в ходе сперматогенеза.
Мутации, вызывающие фенотипическое отсутствие дистрофина, приводят к более тяжелому поражению мышц, чем мутантные аллели DMD, экспрессирующие частично функциональный дистрофии. Корреляции между генотипом и фенотипом для интеллектуального снижения не обнаружено.
Фенотип и развитие мышечной дистрофии Дюшенна
Мужчины с мышечной дистрофией Дюшенна. Миодистрофия Дюшенна — прогрессирующая миопатия, приводящая к дегенерации и слабости мышц. Начинаясь с мышц тазобедренного пояса и сгибателей шеи, мышечная слабость прогрессивно захватывает плечевой пояс и дистальные мышцы конечностей и туловища. Хотя изредка и выявляют случайно больных в период новорожденности за счет гипотонии или задержки развития, обычно больных мальчиков диагностируют в возрасте от 3 до 5 лет при появлении аномалий походки.
К 5 годам большинство пораженных детей используют приемы Говерса и имеют псевдогипертрофию мышц голеней, т.е. увеличение голеней вследствие замены мышц жировой и соединительной тканью. К возрасту 12 лет основная часть больных обездвижены в инвалидном кресле и имеют контрактуры и сколиоз. Большинство пациентов умирают от нарушения легочной функции и пневмонии; средний возраст смерти — 18 лет.
Почти 95% больных миодистрофией Дюшенна имеют те или иные кардиологические отклонения (дилатационная кардиомиопатия или электрокардиографические аномалии), а 84% имеют видимые поражения мышцы сердца при вскрытии. Хронические нарушения сердца бывают почти у 50% пациентов, изредка сердечная недостаточность вызывает у них жалобы. Хотя дистрофии также присутствует в гладких мышцах, гладкомышечные осложнения встречаются редко и включают расширение желудка, заворот кишок и гипотонию мочевого пузыря.
Больные миодистрофией Дюшенна имеют IQ примерно на 1 среднеквадратичное отклонение ниже обычного, и почти треть имеет ту или иную степень умственной отсталости. Причины этого не установлены.

Женщины с мышечной дистрофией Дюшенна
Возраст начала и тяжесть миодистрофии Дюшенна у женщин зависят от степени смещения инактивации Х-хромосомы. Если Х-хромосома, несущая мутантный аллель DMD, активна в большинстве клеток, у женщины развиваются признаки миодистрофии Дюшенна; если преимущественно активна Х-хромосома, несущая нормальный аллель DMD, женщины имеют только несколько или не имеют вообще симптомов данного заболевания.
Независимо от того, есть ли у них клинические симптомы скелетной мышечной слабости, женщины-носительницы имеют отклонения в функции сердечной мышцы, например дилатационную кардиомиопатию, дилатацию левого желудочка и электрокардиографические изменения.
Особенности фенотипических проявлений дистрофии Дюшенна:
• Возраст начала: детство
• Слабость мышц
• Гипертрофия голеней
• Небольшая интеллектуальная недостаточность
• Высокий уровень креатинкиназы сыворотки
Лечение мышечной дистрофии Дюшенна
Диагноз мышечной дистрофии Дюшенна основывается на семейном анамнезе и ДНК-анализе или мышечной биопсии с иммуногистохимическим определением дистрофина.
Глюкокортикоидная терапия может замедлять развитие заболевания в течение нескольких лет. Исследуют несколько видов экспериментального лечения, включая генную передачу. Большинство больных нуждается также в расширенном консультировании, так как имеют дело с психологическими эффектами хронической фатальной болезни.
Риск наследования мышечной дистрофии Дюшенна
Третья часть матерей, родивших единственного больного сына, сами являются носительницами мутаций в гене DMD. Тем не менее определение носительства остается трудной задачей, поскольку в настоящее время доступные молекулярные методы не обнаруживают небольшие мутации типа однонуклеотидных замен. Определение риска носительства в семьях без найденной делеции или дупликации основывается на анализе сцепления, серии анализов уровня креатинкиказы сыворотки крови и мозаичной экспрессии дистрофина в образцах биопсии мышц (из-за случайной инактивации Х-хромосомы). При консультировании в связи с оценкой риска повторения нужно принимать во внимание высокую частоту мозаицизма в половых клетках (приблизительно 14%).
Если мать — носитель, каждый сын имеет 50% риск развития мышечной дистрофии Дюшенна, а каждая дочь — 50% риск унаследовать мутацию DMD. Отражая случайную природу инактивации Х-хромосомы, дочери, унаследовавшие мутацию в гене DMD, имеют низкий риск мышечной дистрофии Дюшенна; тем не менее по причинам, полностью непонятным, риск сердечных аномалий может достигать 50-60%. Если мать не носительница по результатам ДНК-тестирования, у нее остается приблизительно 7% риска родить мальчика с мышечной дистрофией Дюшенна вследствие полового мозаицизма. Для этих матерей показаны генетическое консультирование и, возможно, пренатальная диагностика.
Поскольку анамнез и данные медицинского осмотра, включая повышенный уровень креатинкиназы, позволили предположить миопатию, ребенок для дальнейшего обследования направлен в клинику нейрогенетики. Результаты биопсии мышц показали выраженное изменение размера мышечных волокон, некроз волокон, разрастание жировой и соединительной ткани и отсутствие окрашивания на дистрофии. На основании этих результатов ребенку установлен предварительный диагноз мышечной дистрофии Дюшенна, и он тестирован на делеции в гене дистрофина; оказалось, что у него имеется делеция с 45 по 48 экзон.
Дальнейшее обследование показало, что его мать была носителем этой делеции. Соответственно семье дано заключение, что риск для сыновей составляет 50%, риск для дочерей низкий, но зависит от смещения Х-инактивации, а риск носительства для дочерей равен 50%. Поскольку статус носительницы у матери дает повышенный риск сердечных осложнений, она направлена на кардиологическое обследование.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Прогрессирующая мышечная дистрофия Дюшенна — наследуемая сцеплено с Х-хромосомой патология мышечной системы, проявляющаяся в первые 3-5 лет жизни и характеризующаяся быстро распространяющейся и усугубляющейся мышечной слабостью. Первоначально поражаются мышцы тазового пояса и бедер, затем — плеч и спины, постепенно наступает обездвиженность. Миодистрофия сопровождается скелетными деформациями и поражением сердца. Диагностика дистрофии Дюшенна включает неврологическое и кардиологическое обследование, определение уровня КФК, электромиографию, консультацию генетика, ДНК-анализ, биопсию мышц. Лечение симптоматическое. В связи со слабостью дыхательной мускулатуры на заключительном этапе заболевания требуется ИВЛ.
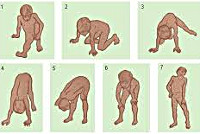
Общие сведения
Прогрессирующая мышечная дистрофия Дюшенна - тяжелая форма миодистрофии, отличающаяся ранним началом, быстрым усугублением мышечной слабости, выраженными деформациями скелета и поражением сердечной мышцы. Впервые была описана французским неврологом Дюшенном в 1853 году. Ее распространенность составляет 1 случай на 4 тыс. новорожденных мальчиков. Патология передается рецессивно сцеплено с Х-хромосомой. Заболевают мальчики. Известны случаи заболевания среди девочек, что связано с кариотипом ХО, гонадотропным мозаицизмом или наличием аномалий в структуре хромосом. Миодистрофия Дюшенна характеризуется началом в первые 3-5 лет жизни ребенка, тяжелым течением, приводящим к полной обездвиженности и гибели пациентов в среднем к возрасту 15-25 лет.
Причины
Развитие мышечной дистрофии Дюшенна связано с наличием мутации в 21-ом локусе короткого плеча Х-хромосомы в гене, кодирующем белок дистрофин. Около 70% случаев болезни вызваны дефектным геном дистрофина, полученным от матери — носительницы патологической мутации. Остальные 30% связаны с появлением свежих мутаций в яйцеклетках матери. В отличие от миодистрофии Беккера, при дистрофии Дюшенна генетические аберрации приводят к сдвигу рамки считывания ДНК и полному прекращению синтеза дистрофина, что и обуславливает более тяжелое течение патологии.
В норме входящий в сарколемму миоцитов дистрофин обеспечивает ее целостность и устойчивость к растяжению, возникающему при сократительной активности мышечных волокон. Отсутствие дистрофина влечет за собой нарушение целостности сарколеммы, разрушение миоцитов и их замещение жировой и соединительной тканью. Клинически этот процесс выражается прогрессирующим снижением способности мышц к сокращению, утратой мышечной силы и тонуса, атрофией мышц.
Симптомы
Дебют миодистрофии Дюшенна приходится на период от 1 до 5 лет. Как правило, уже на 1-ом году жизни заметно некоторое отставание моторного развития ребенка. Отмечается задержка сроков начала сидения, самостоятельного вставания и ходьбы. Когда ребенок начинает ходить, он отличается неуклюжестью и большей, по сравнению со сверстниками, неустойчивостью; часто спотыкается.
Мышечная слабость возникает на 3-4-ом годах жизни. Первоначально она выражается в патологически повышенной утомляемости при ходьбе по лестнице или на длинные расстояния. Со временем становится заметной типичная для миодистрофий утиная походка. Обращают на себя внимание особенности поведения ребенка — каждый раз, поднимаясь из положения сидя на корточках, он активно опирается руками о собственное тело, как бы взбираясь по нему как по лесенке (симптом Говерса).
Прогрессирующая мышечная дистрофия Дюшенна сопровождается нарушениями в костно-суставной системе. Характерны искривление позвоночника (кифоз, усиленный лордоз, сколиоз), деформации грудной клетки (килевидная или седловидная), деформации стоп. Сердечно-сосудистые расстройства обусловлены развитием кардиомиопатии и включают аритмию, лабильность артериального давления, глухость тонов сердца. У 50% больных фиксируются нейроэндокринные расстройства — адипозогенитальная дистрофия, синдром Иценко-Кушинга и др. Около 30% больных страдает олигофренией, как правило, ограничивающейся степенью дебильности. Могут отмечаться СДВГ, расстройства по типу аутизма, дислексия, нарушения краткосрочной памяти.
Осложнения
Уже к 7-10-летнему возрасту дистрофия Дюшенна приводит к выраженным двигательным ограничениям. К 12 годам больные, как правило, утрачивают способность ходить, а к возрасту 15 лет большинство пациентов полностью теряют возможность самостоятельных движений. Распространение дистрофического процесса на дыхательную мускулатуру приводит к прогрессирующему падению жизненной емкости легких (ЖЕЛ) и, в конечном итоге, невозможности совершать дыхательные движения.
Диагностика
Установить диагноз миодистрофии Дюшенна помогает анамнез, неврологическое обследование, результаты электрофизиологического тестирования, определение креатинфосфокиназы (КФК) в биохимическом анализе крови, морфологическое и иммунохимическое исследование образцов мышечной ткани, генетическое консультирование и анализ ДНК:
- ЭФИ. Электронейро- и электромиография определяют сохранность проведения импульсов по нервным волокнам, пониженную амплитуду М-ответа, что свидетельствует о первично-мышечном типе поражения. Характерным является 30-50-кратный подъем уровня креатинфосфокиназы.
- Генетическая диагностика. На консультации генетика проводится генеалогическое исследование, позволяющее выявить наличие случаев миодистрофии Дюшенна в семье больного и определить женщин, являющихся носительницами мутантного гена дистрофина. Диагностика ДНК позволяет выявить аномалии в гене дистрофина. Следует учитывать, что невыявление мутации при ДНК-анализе не говорит о ее отсутствии, поскольку поиск точковых мутаций обычно не входит в задачи анализа из-за его большой длительности и трудоемкости.
- Биопсия. В случаях, когда имеется клиническая картина миодистрофии, а анализ ДНК не выявил наличие мутации, показана биопсия мышц. Морфологическое исследование биоптата определяет разнокалиберность и некроз миоцитов, их замещение соединительнотканными элементами. Иммунохимический анализ говорит о полном отсутствии дистрофина в исследуемых мышечных волокнах.
- Другие исследования. Дополнительно осуществляется обследование костно-мышечной и сердечно-сосудистой систем — проводится консультация ортопеда, рентгенография позвоночника, обзорная рентгенография ОГК, консультация кардиолога, ЭКГ, эхокардиография. По показаниям рекомендуется консультация эндокринолога, пульмонолога и др. специалистов.
При этом дифференциальную диагностику следует проводить с другими миопатиями — метаболической, воспалительной, миодистрофией Беккера, мышечной дистрофией Дрейфуса, дистрофией Эрба-Рота, а также с полиневропатиями, полимиозитом, БАС.
Лечение мышечной дистрофии Дюшенна
Стандартная терапия
Терапия, применяемая в клинической практике, включает симптоматическое и патогенетическое направление. В рамках данных направлений применяется медикаментозная терапия, физическая реабилитация, респираторная поддержка:
- Кортикостероиды. Основная роль в лечении мышечной дистрофии Дюшенна на сегодняшний день отводится глюкокортикостероидам, которые назначаются как способным, так и не способным к самостоятельному передвижению пациентам. ГКС помогают замедлить прогрессирование мышечной слабости, оказывают умеренный пульмопротективный и кардиопротективный эффект, снижают риск развития ортопедических осложнений. Из-а большого количества побочных эффектов глюкокортикостероидной терапии необходим тщательный мониторинг состояния ребенка, своевременная коррекция дозы и схемы приема препарата.
- Метаболическая терапия. Направлена на улучшение обменных процессов в скелетной мускулатуре, костях, сердечной мышце, печени, снижение побочных эффектов от приема ГКС. Включает назначение витаминов группы В, левокарнитина, препаратов Са, витамина D.
- Физическая терапия. С целью минимизировать образование контрактур и продлить двигательную активность пациентов проводится ЛФК, массаж, физиотерапия, пассивная и активная растяжка. Рекомендуется использование ортезов, вертикализатора, специальных шин, занятия лечебным плаванием.
- Респираторная поддержка. Важное значение имеет контроль дыхательной функции и газового состава крови. При падении ЖЕЛ до 40% рекомендована искусственная вентиляция легких в период сна. В дальнейшем время ИВЛ растет пропорционально снижению ЖЕЛ. В начале ИВЛ может осуществляться при помощи масочного аппарата. Затем необходима трахеостомия, и ИВЛ проводится путем присоединения аппарата к трахеостомической трубке. Современные портативные аппараты ИВЛ работают на батареях и могут быть закреплены на инвалидной коляске.
Экспериментальная терапия
Прогноз и профилактика
Из всех форм миодистрофии дистрофия Дюшенна имеет наиболее неблагоприятный прогноз. Манифестация заболевания в раннем возрасте приводит к тому, что к 15 годам пациенты становятся полностью обездвижены. Летальный исход неизбежен. Зачастую больные не достигают 25-летнего возраста. Обычно смертельный исход обусловлен интеркуррентными инфекциями, застойной пневмонией, сердечной или дыхательной недостаточностью.
Профилактические мероприятия направлены на выявление женщин-носительниц аномального гена дистрофина и предупреждение рождения у них больного ребенка. В рамках профилактических мер проводятся консультации генетика для планирующих беременность супружеских пар, консультации беременных и пренатальная ДНК-диагностика.

Фото из архива фонда "Гордей" / Facebook
Как часто и почему возникает это заболевание, почему педиатры нередко путают его с гепатитом, что делать, если вашему ребенку поставлен такой диагноз
Время чтения: 10 мин.
Фото из архива фонда "Гордей" / Facebook
Доктор медицинских наук, президент благотворительного фонда "Гордей" и бабушка Гордея, мальчика с диагнозом миодистрофия Дюшенна, Татьяна Андреевна Гремякова рассказывает о том, что это за болезнь, почему ее часто принимают за гепатит, и что необходимо делать, чтобы дети с этим диагнозом как можно дольше сохраняли активность и жили полной жизнью.
Мышечная дистрофия Дюшенна (МДД) — одно из наиболее распространенных среди редких (орфанных) генетических фатальных нейромышечных заболеваний. В большинстве случаев оно встречается только у мальчиков.
Заболевание связано с мутацией в Х-хромосоме: его частота составляет 1:3500 - 1:5000 новорожденных мальчиков и 1:50 000 000 у новорожденных девочек.
Заболевание развивается вследствие мутации гена, который отвечает за синтез белка дистрофина. У детей прогрессирует повреждение и дегенерация мышц. Со временем мышцы слабеют до такой степени, что дети/подростки не могут самостоятельно передвигаться — в возрасте 10-15 лет мальчики садятся в инвалидную коляску. Параллельно у них развиваются кардиореспираторные нарушения — проблемы с сердцем и дыханием. В возрасте 15-20 лет им уже требуется респираторная поддержка: вначале только ночью, а в дальнейшем – круглосуточно.
Клинические проявления со стороны скелетной мускулатуры и сердца могут различаться в каждом конкретном случае, но смерть, как правило, наступает из-за нарушения функции сердца или дыхания в возрасте 12-25 лет.
Сегодня, благодаря профилактике формирования контрактур, сколиоза и кардиомиопатии, применению стероидов, внедрению респираторной поддержки и другим превентивным мерам, удалось существенно продлить функциональную активность пациентов,
качество и продолжительность их жизни.
Ожидаемая продолжительность жизни мальчиков с МДД, рожденных в последние годы в развитых странах (при условии, что им доступна современная реабилитация, терапия и респираторная поддержка), — 30-40 лет.
Болезнь в большинстве случаев передается от матерей, но сами женщины от нее практически не страдают: они бессимптомные или малосимптомные носители дефектного гена. Ген может передаваться по женской линии многие поколения и никак не проявляться, поэтому рождение ребенка с дистрофией Дюшенна для семьи часто становится неожиданностью. В трети случаев болезнь – результат спонтанной мутации плода без семейной истории.
Ребенок растет, со временем меняется его походка и осанка. Можно заметить, что он ходит на носочках. Ему трудно вставать с пола — поднимается, опираясь на руки (прием Говерса).
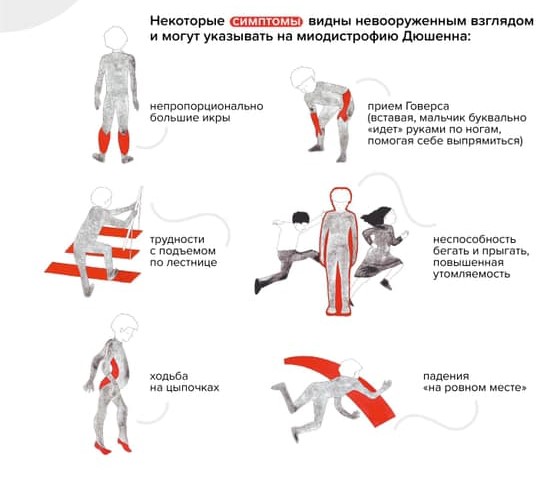
"Прием Говерса" и другие вероятные признаки наличия у ребенка миодистрофии Дюшенна / Фонд "Гордей"
Обращает на себя внимание увеличение икроножных мышц. Позднее трудности при ходьбе нарастают: ребенок с трудом поднимается по лестнице, у него совсем нет сил. В какой-то момент он вовсе перестает ходить и садится в инвалидную коляску.
Если есть сомнения относительно здоровья мальчика, то первое, что нужно сделать, — анализ крови на активность креатинфосфокиназы (КФК). Это фермент, содержащийся в скелетных мышцах, маркер их распада. Обычно при МДД он бывает выше нормы во много раз — например, несколько десятков тысяч единиц при норме в сотню.
МДД — редкое заболевание. Врачи могут ни разу за свою практику не столкнуться с ним и не знать, что делать. Часто ребенку ошибочно ставят диагноз гепатит, перинатальное поражение ЦНС и/или аутизм и назначают лечение, подчас ускоряющее прогрессирование Дюшенна (Рис. 1).
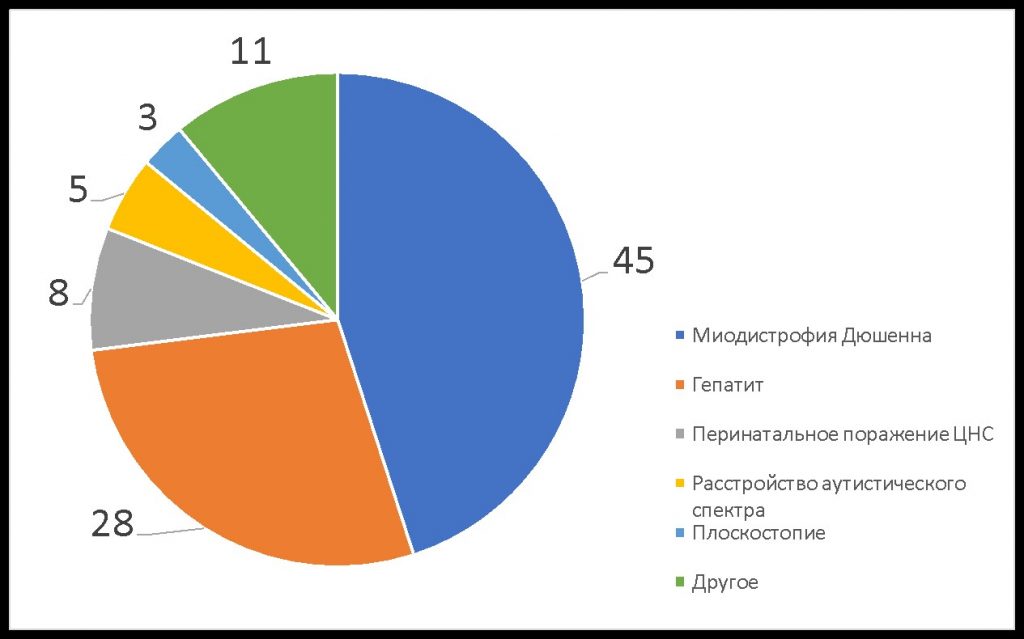
Рис. 1. Результаты опроса ~200 родителей (%): "Какой первый диагноз был поставлен мальчику с миодистрофией Дюшенна в первичном поликлиническом звене?"
Почему гепатит — самый частый из неверных диагнозов? При МДД в крови повышаются уровни трансаминаз, что ошибочно принимается за симптоматику гепатитов. Но эти ферменты в случае МДД мышечного, а не печеночного происхождения.
Если выявлено увеличение уровня КФК выше критического (более 2000 ед), необходимо делать генетический анализ и искать мутацию в гене дистрофина.
Информация для медицинских специалистов
Ген дистрофина — один из самых больших в человеческом организме. Он состоит примерно из 2,5 млн пар нуклеотидов, содержит 79 экзонов, размер гена 2,2 Мб. Уровень мутаций гена относительно высок: в одном из трех случаев МДД вызывается мутацией de novo (впервые возникшее изменение, в отличие от унаследованного – прим.ред). Таким образом, новые случаи возникают даже при наличии хороших инструментов пренатальной диагностики и семейного консультирования для известных случаев.
Высокая частота мутаций также лежит в основе большого разнообразия мутаций, выявленных у пациентов с МДД. Наиболее часто встречаются делеции (~68%), затем по частоте идут дупликации (~11%) одного или нескольких экзонов, реже обнаруживаются небольшие мутации (∼20% пациентов). Мутации могут происходить на любом участке гена, но делеции чаще сконцентрированы между экзонами 45–55, а дупликации — между экзонами 2–10. Тип и расположение мутации определяют ход течения заболевания и то, какое лечение требуется больному.
Сейчас в России применяется специфическая молекулярно-генетическая диагностика повреждения гена дистрофина, определение мутации в каждом конкретном случае.
Генетическое тестирование позволяет точно поставить диагноз. Ранняя и прецизионная диагностика больных МДД необходима для правильного сфокусированного назначения этиопатогенетической терапии. Фактически каждому такому ребенку требуется персонализированная терапия в зависимости от типа и локализации мутации гена.
Бесплатная генетическая диагностика проводится больным с диагнозом МДД, а также бессимптомным пациентам с высокими значениями КФК (более 2000 ЕД\л) с 2019 года. Ограничений по возрасту исследуемых больных нет.
Как попасть на генетическую диагностику?
Только в 30% случаев больной ребенок рождается в результате спонтанной мутации. В 70% случаев носителем мутации является мама ребенка, которая, как правило, не знает об этом.
При планировании беременности женщинам желательно сделать генетический тест на носительство МДД. В некоторых странах такой тест обязателен.
Оценка функциональной активности ребенка позволяет понять его состояние и спрогнозировать дальнейшее течение заболевания. Для этого заболевания нет биохимических маркеров течения болезни. Изменение функциональной и физической активности у детей с МДД выражается в числовых значениях. Это позволяет понять степень выраженности болезни и динамику ее клинической симптоматики.
Если ребенок ходит, его состояние оценивают по методу The North Star (NSAA). Дополнительно оценивается расстояние, пройденное за 6 минут, и время подъема и спуска с четырех ступеней.
Для тех, кто потерял способность ходить, есть метод оценки работы рук — Performance of Upper Limb (PUL) test.
Физическая терапия и реабилитация — основа ухода за больными с МДД на протяжении всей жизни. Она нужна, чтобы поддерживать физическую активность и функциональность.
Современная реабилитация ориентирована:
- на защиту хрупких мышц, на сохранение и поддержание их оптимальной прочности;
- на сведение к минимуму, насколько это возможно, прогрессирования слабости;
- на предотвращение и сведение к минимуму прогрессирующих контрактур и деформаций;
- на поддержку оптимальной кардиореспираторной функции.
На более поздних этапах болезни это:
- использование адаптивного оборудования и вспомогательных технологий (например, коляски, вертикализатора, велотренажера, откашливателя, респираторного оборудования и др.);
- обезболивание;
- поддержка функций и функциональной независимости (поддержание функций рук, обслуживание себя, возможность занятости – работа с компьютером);
- учеба в школе, работа, участие в жизни семьи и общества, а также улучшение качества жизни.

Тутора для ребенка с миодистрофией Дюшенна / Фонд "Гордей"
Для сохранения способности ходить важно следить за постановкой ног ребенка на полную стопу, а также уделять внимание растяжке, особенно мышц голеностопа. Иначе через некоторое время мальчик сможет ходить только на цыпочках, и тогда ему понадобится коляска.
Кроме того, детям с МДД полезны занятия в бассейне с температурой воды 32-34 о С и велосипед (для маленьких детей - велосипед, а когда подросток теряет способность ходить и садится в коляску, тогда – велотренажер для ног и рук).

Занятия в бассейне / Фонд "Гордей"
Если не заниматься физической терапией, ребенок может потерять способность ходить рано и сесть в коляску в 7-8 лет.
В результате начнутся серьезные изменения, в первую очередь, позвоночника: сильный сколиоз, требующий операции на позвоночнике, пострадают сердце и дыхание.
Реабилитация и физическая терапия способны надолго продлить двигательную активность больного.
Позиционирование на стадии коляски фокусируется на том, чтобы сохранить правильное положение тела и ног, не допустить развития сколиоза и контрактур в суставах, сохранить максимальный объем движений рук.
В наиболее тяжелой стадии заболевания важна респираторная реабилитация.
Все о масках для неинвазивной вентиляции легких (НИВЛ) Виды масок, правила подбора, проблемы и осложнения при использовании маски, уход за маской
Медицинская помощь
Миодистрофия Дюшенна — прогрессирующее заболевание, которое со временем поражает многие системы и органы человека. А значит, вести пациента с МДД должна мультидисциплинарная команда врачей под руководством специалиста по нервно-мышечным заболеваниям (например, педиатр или детский невролог).
По мере взросления ребенка эта функция переходит к неврологу, работающему со взрослыми пациентами. Следует заранее определить необходимых специалистов, чья помощь понадобится уже взрослому пациенту.
В современной медицинской и паллиативной концепции ведения пациентов с МДД считается важным ориентироваться на его семью: общаться, координировать уход, информировать об ожидаемых изменениях, связанных с болезнью.
Какие врачи ведут пациента с МДД
| Специалист | Сфера ответственности |
|---|---|
| Невролог | Комплексное ведение болезни на протяжении всей жизни. |
| Реабилитолог | Упреждение развития контрактур и деформации, минимизация боли, продление функциональности и способности к передвижению. |
| Ортопед/Хирург | Поддержка двигательных функций как можно дольше, минимизация контрактур суставов, поддержка позвоночника в прямом положении. |
| Эндокринолог (поддержание костного здоровья) | Уменьшение прогрессирования остеопороза, восстановление при ранних признаках остеопороза. |
| Эндокринолог (рост и половое созревание) | Минимизация нарушений роста, коррекция пубертатного развития, развитие и предотвращение опасного для жизни криза надпочечников. |
| Пульмонолог | Уменьшение респираторных осложнений и сохранение функции дыхательных мышц. |
| Кардиолог | Максимальное продление работы сердца, упреждение формирования сердечной недостаточности и других отклонений. |
| Гастроэнтеролог и диетолог | Профилактика недостаточного или плохого питания, избыточного веса и ожирения. |
| Психолог | Психосоциальная поддержка на протяжении всей жизни, помощь в планировании будущего и формировании представлений о том, как пациенты будут активно участвовать в уходе за собой и ежедневной деятельности. |
| Паллиативная помощь | Симптоматическая терапия, предоставление технических средств реабилитации и медицинских изделий, социальная поддержка. |

Рис. 2. Схема организации мультидисциплинарного ухода за пациентами с миодистрофией Дюшенна на протяжении всей жизни / Фонд "Гордей"
Транзит во взрослую медицину и жизнь
Человек с заболеванием, которое начинается в детском возрасте и ведется детскими врачами, взрослея, переходит во взрослую медицинскую и паллиативную службу. Переход обычно происходит в возрасте от 17 лет до 21 года, в зависимости от системы здравоохранения. Однако, чем старше становится больной с МДД, тем больше он нуждается в медицинской помощи и уходе — ведь болезнь прогрессирует. А значит, без поддержки семьи и медиков не обойтись.
К сожалению, не все больные МДД смогут перейти к самостоятельной и долгой взрослой жизни. Первая причина: около трети молодых взрослых с МДД испытывают психосоциальные трудности или страдают когнитивными нарушениями, которые ограничивают самостоятельность и независимость. Вторая причина: различное течение заболевания.
Обе причины зависят от множественности мутаций гена дистрофина: от чрезвычайно злокачественной, когда дети вовсе не научаются ходить, до доброкачественной, похожей по течению и сроку жизни на миодистрофию Беккера (Birnkrant DJ, Bushby KM, Bann CM, и др., Диагностика и ведение пациентов с мышечной дистрофией Дюшенна, часть 3: первичная помощь, неотложная помощь, психосоциальная помощь и преемственность в оказании помощи на протяжении жизни пациента., The Lancet Neurology, 2018; том 17: 445-55').
И все же для большинства молодых взрослых с МДД следует прогнозировать полноценное участие в планировании будущего и принятии решений.
Ребенку с инвалидностью скоро исполнится 18 лет. Как правильно подготовиться? Статус ребенка-инвалида и инвалида-взрослого сильно различаются. Чтобы не оказаться в трудной ситуации, надо подготовиться к совершеннолетию заранее.
Есть ли лекарство?
Сделать лекарство для МДД сложно: один препарат не вылечит всех, как при СМА. Больные МДД, как снежинки: нет одинаковых — уже описано около десяти тысяч мутаций гена. Поэтому болезнь по-разному проявляется — даже в одной семье у двух братьев.
Какие перспективы?
Кто поможет?

Гордей с мамой, Ольгой Гремяковой. Фото из личного архива / Facebook
Одна из основных задач фонда – информирование и повышение осведомленности о миодистрофии Дюшенна врачебного и родительского сообщества. Чтобы практикующие и будущие педиатры, детские неврологи, медработники детских садов и школ знали о симптомах заболевания, а профильные специалисты имели возможности для обучения, стажировок, обмена опытом и в своей работе опирались на международные стандарты ухода и лучшие мировые практики.
Сообщества по таргетной (подходящей для определенных мутаций) терапии:
Для родителей детей с одной из делеций, корректируемых пропуском экзона 45.
Для родителей детей с одной из делеций, корректируемых пропуском экзона 51.
Для родителей детей с одной из делеций, корректируемых пропуском экзона 53.
С МДД также работают следующие организации:
Федеральные центры, работающие с МДД:
-
ГБОУ ВПО РНИМУ им. Н.И. Пирогова МЗ РФ;
- ФГАУ Национальный медицинский исследовательский Центр Здоровья Детей;
- ФГБУ Центральная Клиническая Больница УДП РФ;
- ФГАОУ ВО РНИМУ им. Н.И. Пирогова Минздрава России – Российская детская клиническая больница;
- ФГБУЗ Центральная клиническая больница РАН;
- ФГБОУ ВО Санкт-Петербургский государственный педиатрический медицинский университет;
- ФГБОУ ВО Сибирский государственный медицинский университет (г.Томск);
- ФГБНУ Томский национальный исследовательский центр РАН.
Читайте также: