
Наследственный ангионевротический отек доклад
Обновлено: 17.05.2024
Наследственный ангионевротический отек (НАО) — хроническое заболевание, относящееся к группе первичных иммунодефицитов с аутосомно-доминантным наследованием и неполной пенетрантностью, связанное с качественным или количественным генетически детерминирова
Наследственный ангионевротический отек (НАО) — хроническое заболевание, относящееся к группе первичных иммунодефицитов с аутосомно-доминантным наследованием и неполной пенетрантностью, связанное с качественным или количественным генетически детерминированным дефектом генов, кодирующих синтез ингибитора эстеразы компонента комплемента C1 (С1inh), которое проявляется в виде рецидивирующих отеков (О.) кожи и слизистых оболочек дыхательных путей, желудочно-кишечного (ЖКТ) и урогенитального трактов [2, 3, 5, 10–14]. Первые упоминания о подобных О. были сделаны Гиппократом в IV в. до н. э.
Первичный дефицит компонентов комплемента встречается редко, так как для манифестации необходимо гомозиготное состояние по аутосомным аллелям. Исключение — делеция гена С1inh при типе 1, точечная мутация при типе 2 (11р11.2-q13). Больные НАО являются гетерозиготами, т. е. имеют один нормальный и один измененный ген, отвечающий за синтез и функционирование С1inh. Частота мутации гена составляет в среднем 1/100 000 [2, 3, 10, 13]. Несмотря на редкое распространение НАО, в некоторых странах созданы национальные общества и регистры, например, во Франции описано 300 случаев [11].
В настоящее время известны 3 различных дефекта [2, 3, 5, 10–12], которые клинически неразличимы:
- первый — в 80–92% случаев количественный дефицит С1inh при определении иммунологическими и энзиматическими методами — НАО I типа;
- второй — в 15% случаев структурный дефект со снижением функциональной активности при нормальном или повышенном уровне С1inh — НАО II типа;
- третий — в 1–5% случаев структурно измененный С1inh (с возможным ↑ концентрации в 3-4 раза); образуются агрегаты с глобулинами (или альбумином) сыворотки крови или активность С1inh блокирована аутоантителами — НАО III типа. Основной механизм ангиоэдемы при НАО связан с эффектами медиаторов — брадикинина, гистамина, производных арахидоновой кислоты, цитокинов.
Критерии диагноза НАО [2, 3, 5, 1-–14, 16, 17] (табл. 1)
Особенности клинической картины
Первые признаки НАО могут возникнуть уже в возрасте нескольких месяцев, но чаще после 1-2 лет жизни. У большинства больных НАО дебют заболевания возникает до 20 лет (60%), гораздо реже — в среднем и даже пожилом возрасте [2, 3, 10, 13]. В пубертатном периоде течение заболевания может утяжелиться в связи с гормональной перестройкой. Больные неоднократно наблюдаются у различных специалистов и не сразу обращаются к аллергологу-иммунологу.
Отсутствие в семейном анамнезе сведений о НАО не исключает возможности постановки подобного диагноза [3, 10, 11, 16]. Клинические проявления у пациентов характеризуются рецидивирующими О. различной локализации: кожи лица (губы, периорбитальная область), шеи, туловища, конечностей, слизистых оболочек верхних отделов дыхательных путей, в том числе гортани, желудочно-кишечного (приступообразные боли в животе) и урогенитального трактов. О. может распространяться на верхние дыхательные пути с вовлечением пищевода, гортани и проявляется дисфагией, дисфонией, симптоматикой обструкции дыхательных путей, напоминающей в ряде случаев бронхиальную астму, и прогрессировать вплоть до асфиксии. Смертность при НАО составляет 20–30% [12]. Больным с О. без симптомов крапивницы и кожного зуда следует уделять особое внимание, так как у них может быть АНО с синдромом недостаточности С1inh, носящий наследственный характер.
Интервалы между обострениями у каждого больного индивидуальны: у некоторых больных О. возникают только после значительной травмы, у других обострения проявляются через каждые 9– 14 дней, вне зависимости от внешних воздействий, на протяжении многих лет [2, 3]. Нередко у больных наблюдается аура в виде слабости, разбитости, мраморность кожи, обильных бледных высыпаний типа кольцевидной эритемы, сохраняющейся во время О., не сопровождающиеся зудом, жжением и ↑ t°. О. может возникать на любом участке кожи или слизистых оболочек.
В более редких случаях при локализации О. на лице могут вовлекаться менингеальные оболочки с проявлением менингеальных симптомов (ригидность затылочных мышц, резкая цефалгия, рвота, иногда судороги), при поражении лабиринтных систем развивается синдром Меньера, что выражается головокружением, тошнотой, рвотой. Все эти симптомы могут встречаться как одновременно, так и по отдельности. При атипичном течении О. могут отсутствовать, также возможны изолированные А., характерны полиартралгии, снижение С4-фракции комплемента. В очень редких случаях описаны: эпилептический приступ, уртикарии, кожная пурпура, феномен Рейно [11]. При лабораторном обследовании больных НАО обнаруживается: С1inh обычно не более 20–30% от нормального (или отсутствие даже в период полной ремиссии), концентрации С2- и С4-компонентов — не более 30–40% от нормальных, уровни С1 и С3 в плазме чаще нормальные. В результате нарушения ингибирования активности С1 постоянно происходит активация комплемента.
Дифференциальная диагностика
Для того чтобы идентифицировать НАО, необходимо провести дифференциальную диагностику генетического, отечного и абдоминального синдромов.
По сравнению с врожденной формой, приобретенный АНО (ПАНО) с дефицитом С1inh, встречается реже, при этом у других членов семьи больного не обнаруживаются аномалии уровня С1inh. Это комплемент-зависимый ПАНО связан с ускорением метаболизма С1inh в 2-3 раза.
Кроме ↓ уровня С4 и С1inh для больных с приобретенной формой заболевания характерно ↓ С1 и С1q, что помогает проводить дифференциальную диагностику. ПАНО можно отличить от НАО по отсутствию дефектности системы комплемента у здоровых родственников и ↓ содержания С1-компонента, при наследственной форме обычно содержащегося в нормальном количестве (табл. 1).
Этиопатогенетическая структура отечного синдрома чрезвычайно вариабельна и имеет полиморфный генез: гипоонкотический, мембраногенный, эндокринный, венозный, лимфогенный О., также отмечены медикаментозные и идиопатические О. [7, 10, с дополнениями].
Распространенные и редкие причины А. могут быть связаны с патологией ЖКТ, гепатобиллиарной системы и поджелудочной железы, системы органов дыхания, сердечно-сосудистой системы, нервной системы, опорно-двигательного аппарата, мочевыделительной системы, половых органов, эндокринного и метаболического генеза, при гематологических заболеваниях и иммунодефицитных состояниях, а также при распространенных инфекционных и паразитарных заболеваниях [12, 14, 16, 18, с дополнениями].
Описанный ниже клинический случай пациентки с направительным диагнозом периодической болезни (ПБ) заставил нас вспомнить об этом заболевании.
Болезнь начинается чаще в детстве и встречается у людей из этнических групп Восточного Средиземноморья (дебют первого приступа ПБ, как правило, наблюдают до 30 лет и в 1,5-2 раза чаще у мужчин с астенической конституцией) — армян, евреев (чаще сефардов), арабов, турков, реже у лиц других национальностей. Средний возраст, в котором дебютируют первые признаки болезни, — 9 лет с колебаниями от 2 мес до 60 лет. Средняя продолжительность болезни — 16,8 лет. Ежегодно отмечается тенденция роста заболеваемости и расширение географии распространения ПБ, причем не только у представителей народов средиземноморского бассейна, но и среди коренного населения: у японцев, русских, болгар, итальянцев. Эндемическим районом считается Армения [4, 5, с дополнениями]. Частота семейных случаев ПБ резко варьирует от 6,8 до 60%. По данным А. А. Айвазяна (n=1036) [1], частота семейных форм составляет 27,5%. Вероятность передачи заболевания от матери к дочери — 2,2%, а от отца к сыну — 28%.
Провоцирующими пароксизмы факторами обычно являются: эмоциональная перегрузка, переутомление, охлаждение, интеркуррентные заболевания, различные пищевые продукты, оперативные вмешательства, перемена климата, обострение язвенной болезни и т. д. Для ПБ характерно обострение с началом менструации, ремиссия болезни во время беременности и после назначения больным прогестерона (табл. 4). Продромальными симптомами (81%) при ПБ являются: слабость, недомогание, зевота, чувство внутренней тревоги, снижение аппетита, раздражительность, ломота в теле, бледность кожных покровов или крапивница, акроцианоз, парестезии, ощущение жжения в животе, полидипсия, похолодание конечностей, озноб и повышение температуры тела, которые обычно наблюдаются за 1-2 ч до развития основного синдрома [4]. По основным клиническим проявлениям выделяют: 1) абдоминальную, 2) торакальную, 3) суставную, 4) лихорадочную и 5) смешанную формы ПБ. Характерны стереотипные периодические пароксизмы высокой t до 38–40°С в 100%, А. в 81,7–98% и/или торакалгий (плевриты и перикардиты) в 33–66%, артралгий и/или артритов в 50–77%, рожеподобной сыпи (эритема) в 46%, лимфаденопатии в 1–6% случаев. Патогномоничные симптомы ПБ (табл. 4) длятся, как правило, от нескольких часов до 3 сут, проходят самостоятельно. Основу клинической картины асептических перитонитов составляет парез ЖКТ. На высоте приступа интенсивные А. сопровождаются тошнотой, рвотой, вздутием живота, задержкой отхождения газов. При объективном исследовании отмечаются: гиперемия лица, тахикардия, вздутие, напряжение, усиление кишечных шумов и болезненность живота, симптомы раздражения брюшины, при этом живот не участвует в акте дыхания. Часто определяется гепатомегалия, реже — спленомегалия.
Указанные симптомы возникают с интервалом несколько недель или месяцев и обычно проявляются в течение 1–3 дней. Пароксизмы рецидивируют с частотой от 1-2 нед до 1 года; могут быть спонтанные ремиссии. Редко наблюдается status periodicus, когда приступы повторяются практически без светлых промежутков; очень редко возможны и абортивные приступы. Имеются наблюдения развития периодической пурпуры, язвенного поражения кожи лица, нейтропении, менингита, психоза, эпилептиформных судорог [4, 6, 9]. Ассоциированный с ПБ; вторичный АА-амилоидоз развивается, по одним данным, в 25%, по другим — в 41,3% случаев, особенно у носителей HLA A28, B5. Поражение почек часто определяет прогноз (причина смерти больных от уремии до 40-летнего возраста) и считается наиболее постоянным и выраженным признаком при ПБ с лихорадкой, абдоминальными и плевральными синдромами. Известны 2 фенотипа ПБ: при первом (чаще) — присоединение амилоидоза происходит при уже имеющейся картине ПБ; при втором (реже) — амилоидоз является первым признаком заболевания. Наряду с этим встречаются случаи ПБ без амилоидоза и случаи, когда амилоидоз служит единственным проявлением заболевания [8].
Описанный клинический случай представляет интерес для врачей различных специальностей с целью проведения дифференциального диагноза НАО и ПБ (табл. 4).
Впервые приступообразные боли в животе (абдоминалгии) появились в возрасте 7 лет. А. продолжительностью более 12 ч повторялись каждые 7–12 дней, сопровождались рвотой, жидким стулом, при этом не отмечалось связи с конкретными причинными факторами, провоцирующими приступы. Неоднократное обследование у различных специалистов, патологии не было выявлено, еженедельные приступы сохранялись. В 13 лет на фоне очередного приступа абдоминалгии госпитализирована в хирургическое отделение с подозрением на холецистит, при лапаротомии изменений желчного пузыря не выявлено, произведена аппендэктомия. Посев на флору серозной жидкости подтвердил асептическое воспаление. С 15 лет на фоне месячных отмечается появление рецидивирующих спонтанных О. различной локализации кожи и слизистых оболочек, исчезающие самостоятельно через 12–16 ч, возникающие еженедельно. О. на коже и слизистых возникали с той же периодичностью, как и приступы А., иногда провоцировались травмой, психоэмоциональной нагрузкой, стоматологическими манипуляциями (экстракция зуба). В 1974 г. впервые О. гортани с удушьем. Применение антигистаминных препаратов и глюкокортикостероидов без положительного эффекта. Со слов пациентки и по данным медицинской документации, лихорадочных реакций, кратковременного повышения температуры тела до, во время и после приступов А. никогда не отмечалось (кроме редких случаев ОРВИ с t до 37,5°С). Больная неоднократно обследовалась у аллерголога, данных, свидетельствующих о наличии аллергопатологии, получено не было, протеинурии не отмечалось. Во время беременности прослеживалась транзиторная протеинурия до 0,03 г/л/сут, умеренное повышение АД до 150 и 90 мм рт. ст., приступы А. продолжались. Роды протекали без развития О. Но после родов О. продолжали возникать через 10–14 дней с той же периодичностью. После родов протеинурия не обнаружена при многократных повторных анализах мочи. После наступления менопаузы абдоминальные боли стали беспокоить реже (1-2 раза в месяц), О. гортани 1-2 раза в год, последний раз в феврале 2001 г.
Учитывая данные анамнеза заболевания, характер клинической картины А., появление АНО без крапивницы, течение заболевания с детского возраста, отсутствие протеинурии, лихорадки и амилоидоза, неэффективность антигистаминных и глюкокортикостероидных препаратов, отрицательный эффект от терапии колхицином и выраженный положительный эффект при назначении Э-АКК на фоне резкого ѓ фракций комплемента — С4, С2, С1inh поставлен следующий диагноз: наследственный ангионевротический отек с количественным дефицитом С1-ингибитора, пароксизмальные абдоминалгии, стадия медикаментозной ремиссии при выписке. Хронический гастродуоденит, вне обострения. Хронический холецистит, вне обострения. Дискенезия желчевыводящих путей. Жировой гепатоз. Хронический панкреатит. Хронический колит. Лямблиоз кишечника. Артериальная гипертензия II степени. Хроническая венозная недостаточность 0-I ст. Ожирение II степени. Остеохондроз поясничного отдела позвоночника с корешковым синдромом, вне обострения. Вегетативно-сосудистая дистония с явлениями церебральной вазопатии. Ангиодистония сетчатки. Пациентке выдан паспорт, подтверждающий диагноз НАО. Проведена противопротозойная терапия (макмирор, тинидазол), полоскание горла антисептиками, энзимотерапия (мезим-форте), назначена гипотензивная терапия арифоном. За время пребывания в стационаре больная отмечала приступ болей в правом подреберье, чувство дискомфорта в грудной клетке, было проведено обследование, данных, свидетельствующих о наличии острого панкреатита, холецистита на УЗИ, не получено, приступ купирован приемом анальгетиков, в/в введением Э-АКК. Начата базисная терапия дановалом в суточной дозе 600 мг. В дальнейшем состояние больной стабилизировалось, самочувствие улучшилось. О. слизистых и кожи нет. Дыхание и гемодинамика в норме. Рекомендовано продолжить прием дановала 600 мг/сут и завершить коррекцию выявленных нарушений, а также обследование всех родственников. Пациентка более 12 мес находится на терапии дановалом в дозировке сначала 600 мг/сут, далее через 3 мес по 200 мг/сут. О. кожи и слизистых не отмечается.
Рекомендации по лечению и ведению пациентов при НАО [2–3, 10–12, 14]
Пациенты с НАО требуют не только дифференцированного индивидуального подхода, но и назначения препаратов, учитывая возможный риск, изменение качества жизни. Такие предпосылки обусловлены тем, что дебют заболевания чаще происходит в пубертатный период, когда имеются не только эндокринные, но и психологические проблемы у пациентов с НАО.
В случае дебюта НАО у девочек в пубертатном периоде или женщин во время беременности и лактации рекомендуется начинать лечение с введения нативной плазмы.
Для многих пациентов зрелого возраста характерно многолетнее и безуспешное лечение у врачей различных специальностей — терапевтов, гастроэнтерологов, гинекологов, хирургов, врачей приемных комиссий военкоматов и др. В связи с этим больным НАО часто ставят неправильный диагноз (О. Квинке, крапивница, аллергия, острый живот, острый аппендицит, острый холецистит, стеноз чревного ствола, ангина, ревматологическое заболевание, кровоизлияние в мозг, мигрень, эпилепсия и др.) и назначают неадекватную для данного заболевания терапию, что становится причиной высокой смертности таких пациентов. Около 25% больных умирают от О. гортани в возрасте до 30 лет. Многим больным неоднократно проводят лапаротомии и трахеотомии из-за некупирующихся О. [2, 3, 10, 13].
Данный клинический случай ярко демонстрирует сложности дифференциальной диагностики абдоминального синдрома при НАО и ПБ, что отражено в таблице 4. Это особенно важно для практикующих врачей-хирургов, терапевтов, врачей скорой помощи, оториноларингологов, аллергологов. От клиницистов требуется не только сбор анамнеза заболевания и владение методами объективного исследования, но и знание клинической картины кожно-висцерального синдрома — ПБ и атипичных проявлений НАО, ставших актуальной проблемой современной клинической аллергологии и иммунологии.
По вопросам литературы обращайтесь в редакцию.
Г. Х. Викулов
Е. С. Феденко, доктор медицинских наук
Т. В. Латышева, доктор медицинских наук, профессор ГНЦ Институт иммунологии МЗ РФ, г. Москва
Наследственный ангиоотек, НАО, ангиоотек, абдоминальная атака, дефект системы комплемента, система комплемента, брадикинин, С1-ингибитор, долгосрочная профилактика, краткосрочная профилактика, купирование отека.
Термины и определения
Ангиоотек – локализованный транзиторно остро возникающий, склонный к рецидивированию отек кожи или слизистых оболочек. Обычно длится от 2ч до нескольких дней и проходит самостоятельно. Этиологические факторы ангиоотёков различны.
Список сокращений
АД – артериальное давление
и АПФ- ингибиторы ангиотензинпревращающего фермента
ЖКТ – желудочно-кишечный тракт
ЛС – лекарственные средства
МКБ -10 Международная статистическая классификация болезней и проблем, связанных со здоровьем, 10-го пересмотра, принятая 43-ей Всемирной Ассамблеей Здравоохранения
НАО – наследственный ангиоотёк
НПВП - нестероидные противовоспалительные препараты
ПАО – приобретенный ангиоотёк
СОЭ – скорость оседания эритроцитов
СРБ – С-реактивный белок
Т4 – тироксин свободный
ЧСС – частота сердечных сокращений
С1-ингибитор – ингибитор первого компонента комплемента
С1-INH95 Kd - C1-ингибитор низкомолекулярной массы с весом 95 килодальтон
С1q – фактор первого компонента комплемента
С4 – компонент комплемента
IgE – иммуноглобулин класса Е
IgG – иммуноглобулин класса G
IgМ – иммуноглобулин класса М
Ф XII – коагуляционный фактор I
Термины и определения
Ангиоотек – локализованный транзиторно остро возникающий, склонный к рецидивированию отек кожи или слизистых оболочек. Обычно длится от 2ч до нескольких дней и проходит самостоятельно. Этиологические факторы ангиоотёков различны.
1. Краткая информация
1.1 Определение
Наследственный ангиоотек (НАО) – редкое, потенциально жизнеугрожающее генетически детерминированное заболевание, связанное с дефицитом или снижением функции С1-ингибитора.[1,2] Основными проявлениями НАО являются рецидивирующие отеки глубоких слоев дермы различной локализации, которые сохраняются от нескольких часов до нескольких дней. Характерными особенностями отеков при НАО являются: отсутствие зуда, гиперемии кожи, сопутствующей крапивницы, а также отсутствие эффекта от лечения ГКС и антигистаминными средствами. [1,2,3,4]
1.2 Этиология и патогенез
НАО относится к первичным иммунодефицитам без инфекционного синдрома, в патогенезе данного заболевания основную роль играют нарушения в системе комплемента. [2]
Ключевую роль в развитии отеков играет генетически обусловленный дефицит С1-ингибитора, приводящий к нарушению ингибирования С1r и C1s компонентов комплемента, а также аутоактивации фактора Хагемана системы свертывания крови. Следствием этого является усиленное образование брадикинина из кининогена. Накопление вазоактивного вещества – брадикинина, в свою очередь, приводит к развитию обратимого увеличения проницаемости эндотелия (рис. 1). [6,8]
Рис. 1. Точки приложения С1-ингибитора. С1-ингибитор предотвращает переход прекалликреина в каллекреин, плазминогена в плазмин, активацию ХII фактора свертывающей системы крови. При недостатке С1-ингибитора происходит накопление брадикинина. Брадикинин увеличивает проницаемость сосудистой стенки, вызывает экстравазацию, в конечном итоге развивается отек.
1.3 Эпидемиология
Распространенность 1:50 000. [4,5,6]
Для данного заболевания в большинстве случаев характерен аутосомно-доминантный тип наследования. Около 25% не имеют семейной истории ангиоотеков (мутации de novo).[5,6,7]
1.4 Кодирование по МКБ 10
D 84.1 – дефект в системе комплемента.
1.5 Классификация
Комментарии. Отмечается дефицит С1-ингибитора в плазме, обусловленный нефункционирующим геном. При этом уровень С1-ингибитора может варьировать от неопределяемого до снижения на 30% от нормального значения. [4,8]
Комментарии. Уровень С1-ингибитора в пределах нормы или повышен, отмечается снижение функциональной активности С1-ингибитора. [4,8]
- НАО 3-го типа (редко встречающийся тип НАО, распространенность неизвестна).
Комментарии. НАО с нормальным уровнем С1-ингибитора (эстрогензависимый). Считается, что развитие ангиоотеков при НАО III типа связано с генетическим нарушением контроля ХII фактора свертывания крови. Отличительной особенностью НАО 3-го типа является нормальный уровень С1-ингибитора и его функциональной активности. Клинические симптомы идентичны таковым при первых двух типах наследственных ангиоотеков. Его особенностью является зависимость симптомов от высокого уровня эстрогенов, и, соответственно, для него характерны обострения во время беременности, при применении пероральных контрацептивов или заместительной терапии эстрогенами при лечении климактерического синдрома. Преимущественно болеют женщины. Основанием постановки диагноза служит типичная клиническая картина заболевания в сочетании с выявлением мутаций в гене XII фактора свертывания крови и/или наличием семейного анамнеза. [6,8,9]
2. Диагностика
Диагноз НАО ставится на основании данных анамнеза, особенностей клинической картины, результатов физикального и лабораторного обследования. [7]
2.1 Жалобы и анамнез
В пользу наследственного ангиоотека свидетельствуют:
2.2 Физикальное обследование
Важным диагностическим критерием является характер отека, связанный с участием брадикинина в его формировании: отек бледный и не зудящий, плотный (при надавливании на него не остается ямки).
- Возможно наличие покалывания, жжения, болезненности в месте отека; [3,11]
- При абдоминальных отеках у больного выявляют отек участка кишки и асцитический выпот; [8,9,10]
- При развитии потенциально фатального отека гортани – осиплость голоса, дисфония, одышка, стридор; [9,10]
- При отеке языка отмечается существенное увеличение его в объеме, часто язык не помещается в ротовой полости. При отеке мочевыводящей путей возникает задержка мочи; [8,9,10]
- Крапивница отсутствует, возможно наличие маргинальной эритемы; [3,9,11]
- Сильные головные боли наблюдаются при отеке мозговых оболочек; [8]
- Осмотр доступных обследованию верхних дыхательных путей, оценка звучности голоса, возможности глотания для исключения развивающегося и угрожающего жизни отека ротоглоточной области; [8]
- Тщательный общий осмотр для выявления лимфоаденопатии, спленомегалии, артропатии и другой патологии, возможно, являющейся причиной других форм АО. [8]
2.3 Лабораторные исследования
Для подтверждения диагноза НАО рекомендовано определение следующих параметров (таблица 1):
- исследовать уровни С4 компонента системы комплемента;
- определить уровень и функциональную активность С1-ингибитора;
- определить наличие антител к С1 ингибитору;
- генетическое исследование. [8,9,12]
Уровень убедительности рекомендаций B.
- При подозрении на ПАО рекомендовано дальнейшее обследование с определением С1q и С1-INH95 Kd.
Уровень убедительности рекомендаций С.
Комментарии. Концентрация С1q-компонента, как правило, снижена при ПАО (таблица 1).[8,9,10]
Таблица 1. Лабораторная диагностика наследственного и приобретенного ангиоотека
Результаты лабораторных исследований при наследственном ангиоотеке (НАО) и приобретенном ангиоотеке (ПАО)
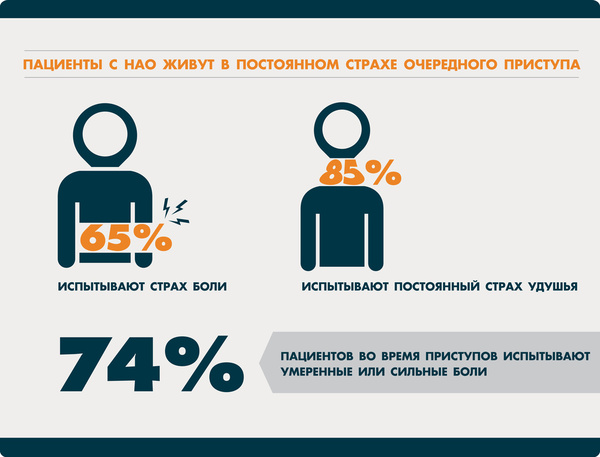
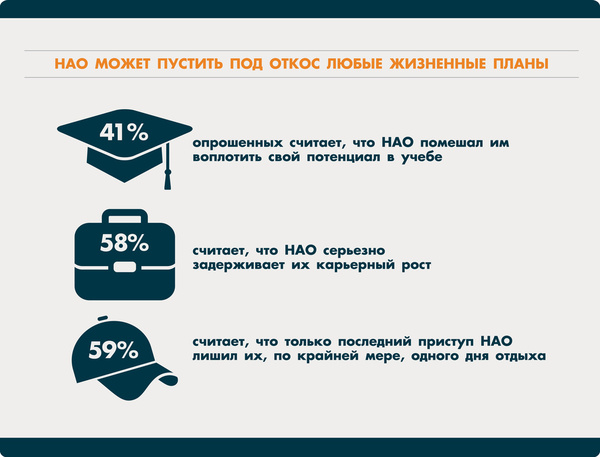
Наследственный ангионевротичекий отёк - редкое, жизнеугрожающее заболевание, которое относится к группе первичных иммунодефицитов. Причина - недостаточность общего уровня или снижение функциональной активности С1-ингибитора системы комплемента. Жизнь таких больных становится кошмаром: они никогда не знают, где и когда начнется отек. Пациенты нередко испытывают страх очередного приступа, для них характерны чувство одиночества, ощущение безысходности и бесконечные проблемы на работе, в учебе и быту.


Наследственный ангионевротический отек (НАО) — хроническое заболевание, относящееся к группе первичных иммунодефицитов с аутосомно-доминантным наследованием и неполной пенетрантностью, связанное с качественным или количественным генетически детерминированным дефектом генов, кодирующих синтез ингибитора эстеразы компонента комплемента C1 (С1inh), которое проявляется в виде рецидивирующих отеков (О.) кожи и слизистых оболочек дыхательных путей, желудочно-кишечного (ЖКТ) и урогенитального трактов [2, 3, 5, 10–14]. Первые упоминания о подобных отёках были сделаны Гиппократом в IV в. до н. э.
Отсутствие в семейном анамнезе сведений о НАО не исключает возможности постановки подобного диагноза [3, 10, 11, 16]. Клинические проявления у пациентов характеризуются рецидивирующими О. различной локализации: кожи лица (губы, периорбитальная область), шеи, туловища, конечностей, слизистых оболочек верхних отделов дыхательных путей, в том числе гортани, желудочно-кишечного (приступообразные боли в животе) и урогенитального трактов. О. может распространяться на верхние дыхательные пути с вовлечением пищевода, гортани и проявляется дисфагией, дисфонией, симптоматикой обструкции дыхательных путей, напоминающей в ряде случаев бронхиальную астму, и прогрессировать вплоть до асфиксии. Смертность при НАО составляет 20–30% [12]. Больным с О. без симптомов крапивницы и кожного зуда следует уделять особое внимание, так как у них может быть АНО с синдромом недостаточности С1inh, носящий наследственный характер.
Интервалы между обострениями у каждого больного индивидуальны: у некоторых больных О. возникают только после значительной травмы, у других обострения проявляются через каждые 9– 14 дней, вне зависимости от внешних воздействий, на протяжении многих лет [2, 3]. Нередко у больных наблюдается аура в виде слабости, разбитости, мраморность кожи, обильных бледных высыпаний типа кольцевидной эритемы, сохраняющейся во время О., не сопровождающиеся зудом, жжением и ↑ t°. О. может возникать на любом участке кожи или слизистых оболочек.
Распространенные и редкие причины А. могут быть связаны с патологией ЖКТ, гепатобиллиарной системы и поджелудочной железы, системы органов дыхания, сердечно-сосудистой системы, нервной системы, опорно-двигательного аппарата, мочевыделительной системы, половых органов, эндокринного и метаболического генеза, при гематологических заболеваниях и иммунодефицитных состояниях, а также при распространенных инфекционных и паразитарных заболеваниях [12, 14, 16, 18, с дополнениями].
Болезнь начинается чаще в детстве и встречается у людей из этнических групп Восточного Средиземноморья (дебют первого приступа ПБ, как правило, наблюдают до 30 лет и в 1,5-2 раза чаще у мужчин с астенической конституцией) — армян, евреев (чаще сефардов), арабов, турков, реже у лиц других национальностей. Средний возраст, в котором дебютируют первые признаки болезни, — 9 лет с колебаниями от 2 мес до 60 лет. Средняя продолжительность болезни — 16,8 лет. Ежегодно отмечается тенденция роста заболеваемости и расширение географии распространения ПБ, причем не только у представителей народов средиземноморского бассейна, но и среди коренного населения: у японцев, русских, болгар, итальянцев. Эндемическим районом считается Армения [4, 5, с дополнениями]. Частота семейных случаев ПБ резко варьирует от 6,8 до 60%. По данным А. А. Айвазяна (n=1036) [1], частота семейных форм составляет 27,5%. Вероятность передачи заболевания от матери к дочери — 2,2%, а от отца к сыну — 28%.
Провоцирующими пароксизмы факторами обычно являются: эмоциональная перегрузка, переутомление, охлаждение, интеркуррентные заболевания, различные пищевые продукты, оперативные вмешательства, перемена климата, обострение язвенной болезни и т. д. Для ПБ характерно обострение с началом менструации, ремиссия болезни во время беременности и после назначения больным прогестерона. Продромальными симптомами (81%) при ПБ являются: слабость, недомогание, зевота, чувство внутренней тревоги, снижение аппетита, раздражительность, ломота в теле, бледность кожных покровов или крапивница, акроцианоз, парестезии, ощущение жжения в животе, полидипсия, похолодание конечностей, озноб и повышение температуры тела, которые обычно наблюдаются за 1-2 ч до развития основного синдрома [4]. По основным клиническим проявлениям выделяют: 1) абдоминальную, 2) торакальную, 3) суставную, 4) лихорадочную и 5) смешанную формы ПБ. Характерны стереотипные периодические пароксизмы высокой t до 38–40°С в 100%, А. в 81,7–98% и/или торакалгий (плевриты и перикардиты) в 33–66%, артралгий и/или артритов в 50–77%, рожеподобной сыпи (эритема) в 46%, лимфаденопатии в 1–6% случаев. Патогномоничные симптомы ПБ длятся, как правило, от нескольких часов до 3 сут, проходят самостоятельно. Основу клинической картины асептических перитонитов составляет парез ЖКТ. На высоте приступа интенсивные А. сопровождаются тошнотой, рвотой, вздутием живота, задержкой отхождения газов. При объективном исследовании отмечаются: гиперемия лица, тахикардия, вздутие, напряжение, усиление кишечных шумов и болезненность живота, симптомы раздражения брюшины, при этом живот не участвует в акте дыхания. Часто определяется гепатомегалия, реже — спленомегалия.
Указанные симптомы возникают с интервалом несколько недель или месяцев и обычно проявляются в течение 1–3 дней. Пароксизмы рецидивируют с частотой от 1-2 нед до 1 года; могут быть спонтанные ремиссии. Редко наблюдается status periodicus, когда приступы повторяются практически без светлых промежутков; очень редко возможны и абортивные приступы. Имеются наблюдения развития периодической пурпуры, язвенного поражения кожи лица, нейтропении, менингита, психоза, эпилептиформных судорог [4, 6, 9]. Ассоциированный с ПБ; вторичный АА-амилоидоз развивается, по одним данным, в 25%, по другим — в 41,3% случаев, особенно у носителей HLA A28, B5. Поражение почек часто определяет прогноз (причина смерти больных от уремии до 40-летнего возраста) и считается наиболее постоянным и выраженным признаком при ПБ с лихорадкой, абдоминальными и плевральными синдромами. Известны 2 фенотипа ПБ: при первом (чаще) — присоединение амилоидоза происходит при уже имеющейся картине ПБ; при втором (реже) — амилоидоз является первым признаком заболевания. Наряду с этим встречаются случаи ПБ без амилоидоза и случаи, когда амилоидоз служит единственным проявлением заболевания [8].
Описанный клинический случай представляет интерес для врачей различных специальностей с целью проведения дифференциального диагноза НАО и ПБ.
Впервые приступообразные боли в животе (абдоминалгии) появились в возрасте 7 лет. А. продолжительностью более 12 ч повторялись каждые 7–12 дней, сопровождались рвотой, жидким стулом, при этом не отмечалось связи с конкретными причинными факторами, провоцирующими приступы. Неоднократное обследование у различных специалистов, патологии не было выявлено, еженедельные приступы сохранялись. В 13 лет на фоне очередного приступа абдоминалгии госпитализирована в хирургическое отделение с подозрением на холецистит, при лапаротомии изменений желчного пузыря не выявлено, произведена аппендэктомия. Посев на флору серозной жидкости подтвердил асептическое воспаление. С 15 лет на фоне месячных отмечается появление рецидивирующих спонтанных О. различной локализации кожи и слизистых оболочек, исчезающие самостоятельно через 12–16 ч, возникающие еженедельно. О. на коже и слизистых возникали с той же периодичностью, как и приступы А., иногда провоцировались травмой, психоэмоциональной нагрузкой, стоматологическими манипуляциями (экстракция зуба). В 1974 г. впервые О. гортани с удушьем. Применение антигистаминных препаратов и глюкокортикостероидов без положительного эффекта. Со слов пациентки и по данным медицинской документации, лихорадочных реакций, кратковременного повышения температуры тела до, во время и после приступов А. никогда не отмечалось (кроме редких случаев ОРВИ с t до 37,5°С). Больная неоднократно обследовалась у аллерголога, данных, свидетельствующих о наличии аллергопатологии, получено не было, протеинурии не отмечалось. Во время беременности прослеживалась транзиторная протеинурия до 0,03 г/л/сут, умеренное повышение АД до 150 и 90 мм рт. ст., приступы А. продолжались. Роды протекали без развития О. Но после родов О. продолжали возникать через 10–14 дней с той же периодичностью. После родов протеинурия не обнаружена при многократных повторных анализах мочи. После наступления менопаузы абдоминальные боли стали беспокоить реже (1-2 раза в месяц), О. гортани 1-2 раза в год, последний раз в феврале 2001 г.
Учитывая данные анамнеза заболевания, характер клинической картины А., появление АНО без крапивницы, течение заболевания с детского возраста, отсутствие протеинурии, лихорадки и амилоидоза, неэффективность антигистаминных и глюкокортикостероидных препаратов, отрицательный эффект от терапии колхицином и выраженный положительный эффект при назначении Э-АКК на фоне резкого ѓ фракций комплемента — С4, С2, С1inh поставлен следующий диагноз: наследственный ангионевротический отек с количественным дефицитом С1-ингибитора, пароксизмальные абдоминалгии, стадия медикаментозной ремиссии при выписке. Хронический гастродуоденит, вне обострения. Хронический холецистит, вне обострения. Дискенезия желчевыводящих путей. Жировой гепатоз. Хронический панкреатит. Хронический колит. Лямблиоз кишечника. Артериальная гипертензия II степени. Хроническая венозная недостаточность 0-I ст. Ожирение II степени. Остеохондроз поясничного отдела позвоночника с корешковым синдромом, вне обострения. Вегетативно-сосудистая дистония с явлениями церебральной вазопатии. Ангиодистония сетчатки. Пациентке выдан паспорт, подтверждающий диагноз НАО. Проведена противопротозойная терапия (макмирор, тинидазол), полоскание горла антисептиками, энзимотерапия (мезим-форте), назначена гипотензивная терапия арифоном. За время пребывания в стационаре больная отмечала приступ болей в правом подреберье, чувство дискомфорта в грудной клетке, было проведено обследование, данных, свидетельствующих о наличии острого панкреатита, холецистита на УЗИ, не получено, приступ купирован приемом анальгетиков, в/в введением Э-АКК. Начата базисная терапия дановалом в суточной дозе 600 мг. В дальнейшем состояние больной стабилизировалось, самочувствие улучшилось. О. слизистых и кожи нет. Дыхание и гемодинамика в норме. Рекомендовано продолжить прием дановала 600 мг/сут и завершить коррекцию выявленных нарушений, а также обследование всех родственников. Пациентка более 12 мес находится на терапии дановалом в дозировке сначала 600 мг/сут, далее через 3 мес по 200 мг/сут. О. кожи и слизистых не отмечается. Рекомендации по лечению и ведению пациентов при НАО [2–3, 10–12, 14]
Пациенты с НАО требуют не только дифференцированного индивидуального подхода, но и назначения препаратов, учитывая возможный риск, изменение качества жизни. Такие предпосылки обусловлены тем, что дебют заболевания чаще происходит в пубертатный период, когда имеются не только эндокринные, но и психологические проблемы у пациентов с НАО.
Данный клинический случай ярко демонстрирует сложности дифференциальной диагностики абдоминального синдрома при НАО и ПБ. Это особенно важно для практикующих врачей-хирургов, терапевтов, врачей скорой помощи, оториноларингологов, аллергологов. От клиницистов требуется не только сбор анамнеза заболевания и владение методами объективного исследования, но и знание клинической картины кожно-висцерального синдрома — ПБ и атипичных проявлений НАО, ставших актуальной проблемой современной клинической аллергологии и иммунологии.
Г. Х. Викулов Е. С. Феденко, доктор медицинских наук Т. В. Латышева, доктор медицинских наук, профессор ГНЦ Институт иммунологии МЗ РФ, г. Москва
Другие статьи
Колонка эксперта - Беллы Брагвадзе. Удивительный мир иммунитета.
Чем настоящий иммунодефицит отличается от частой простуды
Почему дети должны болеть, о бессмысленности иммуномодуляторов и о том, чем настоящий иммунодефицит отличается от частой простуды, — иммунолог Анна Щербина Иммунодефицит — это состояние, сопровождающееся значительными и долговременными изменениями в иммунной системе и серьезными симптомами. Есть иммунодефициты вторичные, а есть первичные (ПИД). Первичные обусловлены генетически. Как правило, симптомы возникают в раннем возрасте, однако иногда могут возникнуть и у взрослых. Но в любом случае проявления будут очень тяжелыми. С первичными иммунодефицитами встречаются крайне редко. Подтвердить многие такие заболевания можно, обнаружив дефект гена. Но пока, правда, найдены мутации не при всех ПИД, поиск продолжается. Текст: Дарья Саркисян Фотографии: Максим Шер Журнал "Большой город"
ЧТО ТАКОЕ ПЕРВИЧНЫЙ ИММУНОДЕФИЦИТ
Как рассказать детям об иммунитете
Презентация для школьников о том, что такое иммунитет, какие нарушения встречаются, как живут дети с первичным иммунодефицитом и как им можно помочь.
Первичный иммунодефицит. Х-сцепленный лимфопролиферативный синдром
Х-сцепленный лимфопролиферативный синдром – первичный иммунодефицит, при котором у пациентов мужского пола отмечается нарушение иммунного ответа на вирус Эпштейн-Барра.
Первичный иммунодефицит. Аутоиммунный лимфопролиферативный синдром
Аутоиммунный лимфопролиферативный синдром – первичный иммунодефицит, при котором отмечается хроническое незлокачественное увеличение лимфоузлов, печени и селезенки, аутоиммунная патология, повышение уровня иммуноглобулинов в крови.
Первичный иммунодефицит. Синдром Ди Джорджи
Синдром Ди Джорджи – врожденный дефект, который приводит к гипоплазии или отсутствию тимуса (вилочковой железы) в сочетании с пороками развития крупных сосудов, сердца, паращитовидных желез, костей лицевого черепа и верхних конечностей
Оптимизация диагностики и терапии наследственного ангионевротического отека у взрослых.
Особенности редкой формы первичного иммунодефицита, клинические проявления, иммунологические нарушения и принципы терапии наследственного ангионевротического отека. Индивидуальные планы самоконтроля для каждого пациента и оценкаа их эффективность. Караулов А.В., Сидоренко И.В., Капустина А.С. Первый Московский государственный медицинский университет им. И. М. Сеченова, Москва
Первичный иммунодефицит. Хроническая гранулематозная болезнь
Хроническая гранулематозная болезнь (ХГБ) – генетическое заболевание, связанное с дефектом фагоцитов, клеток иммунной системы, которые защищают организм путем поглощения (фагоцитоза) вредных чужеродных частиц, бактерий, а также мертвых или погибающих клеток, из-за которого снижается их антимикробная активность.
Иммунодефицитные состояния
Первичный иммунодефицит. ОВИН - общая вариабельная иммунная недостаточность
Общая вариабельная иммунная недостаточность - нарушение, характеризующееся низкими уровнями иммуноглобулинов (антител) в сыворотке крови и повышенной чувствительностью к инфекциям. Данная статья предназначена для пациентов и членов их семей и не должна заменять совета клинициста-иммунолога.
Первичный иммунодефицит. Синдром Вискотта-Олдрича
Синдром Вискотта-Олдрича является первичным иммунодефицитным состоянием, поражающим как Т- лимфоциты, так и В-лимфоциты. Также тяжело поражаются тромбоциты - клетки, помогающие останавливать кровотечение. Информация в статье предназначена для пациентов и членов их семей и не должна заменять рекомендаций и назначений лечащего врача и клинициста-иммунолога.
Первичный иммунодефицит. Х-сцепленная агаммаглобулинемия
У больных с Х-сцепленной агаммаглобулинемией основным дефектом является неспособность предшественников В-лимфоцитов созревать до состояния В-лимфоцитов, а затем плазматических клеток. Поскольку у этих больных нет клеток, вырабатывающих иммуноглобулины, наступает тяжелая недостаточность иммуноглобулинов. Информация в статье предназначена для пациентов и членов их семей и не должна заменять рекомендаций и назначений лечащего врача и клинициста-иммунолога.
Первичный иммунодефицит. ТКИН - тяжелая комбинированная иммунная недостаточность
Тяжелая комбинированная иммунная недостаточность (ТКИН) - самый тяжелый диагноз в списке первичных иммунодефицитов - является редким синдромом, обусловленным различными генетическими факторами, и сочетающим отсутствие функций Т- и В- лимфоцитов (а во многих случаях также отсутствие функции естественных киллеров или NK-лимфоцитов). Эти нарушения приводят к чрезвычайной чувствительности к тяжелым инфекциям. Информация в статье предназначена для пациентов и членов их семей и не должна заменять рекомендаций и назначений лечащего врача и клинициста-иммунолога.
12 настораживающих признаков первичного иммунодефицита
ПИД не СПИД. Первичный иммунодефицит является врожденным нарушением в иммунной системе, имеющим генетическую природу. Показанием для направления к иммунологу является сочетание рецидивирующих вирусных и бактериальных инфекций либо наличие тяжелых, затяжных бактериальных инфекций. Данные Всемирной организации здравоохранения свидетельствуют о том, что частота ОРВИ 8 раз в год является нормальным показателем для детей дошкольного и младшего школьного возраста, посещающих детские учреждения.
Часто болеющие дети: чем они больны на самом деле?
Инфекции уха, горла, носа, а также бронхолёгочные инфекции составляют основной перечень заболеваний в детском возрасте. Данные ВОЗ свидетельствуют о том, что частота ОРВИ 8 раз в год является нормальным показателем для детей дошкольного и младшего школьного возраста, посещающих детские учреждения. Показанием для направления к иммунологу является сочетание рецидивирующих вирусных и бактериальных инфекций либо наличие тяжелых, затяжных бактериальных инфекций.
Иммунодефициты у детей.
Диагностика семей с иммунодефицитом
Первичные иммунодефициты, являются наследуемыми заболеваниями, при которых родители являются носителями больного гена и передают его детям. В результате чего у ребенка развивается заболевание. В настоящее время в связи с развитием генетики и иммунологии известны многие гены, мутация в которых приводит к развитию различных форм первичных иммунодефицитов.
Изолированные рецидивирующие ангиоотеки представляют собой важную клиническую проблему, актуальность которой обусловлена не только недостаточными сведениями о механизмах развития, но и серьезным влиянием болезни на все стороны жизни пациентов, трудностями диагностики и лечения данного заболевания [1]. В обзоре дана классификация ангиоотеков, освещены вопросы этиологии, патогенеза, клиники, диагностики и лечения наследственных и приобретенных комплементзависимых ангиоотеков.
Изолированные рецидивирующие ангиоотеки представляют собой важную клиническую проблему, актуальность которой обусловлена не только недостаточными сведениями о механизмах развития, но и серьезным влиянием болезни на все стороны жизни пациентов, трудностями диагностики и лечения данного заболевания [1]. В обзоре дана классификация ангиоотеков, освещены вопросы этиологии, патогенеза, клиники, диагностики и лечения наследственных и приобретенных комплементзависимых ангиоотеков.
Таблица 4. Новые лекарственные препараты для лечения наследственных отеков (по данным клинических исследований)
Ангиоотек (АО) – это результат локального повышения проницаемости подслизистых и подкожных капилляров и венул, клинически характеризуется локализованным внезапно возникающим транзиторным и часто рецидивирующим отеком кожи или слизистых оболочек. Ангиоотек может возникать в сочетании с явлениями крапивницы или протекать в изолированном варианте, не сопровождаясь образованием волдырей и зудом.
В настоящее время в зависимости от классификационного признака ангиоотеки подразделяются следующим образом [4].
I. По клинической характеристике.
1. По течению: острый (до 6 недель) и хронический (более 6 недель).
2. По сочетанию с крапивницей: изолированный и сочетанный.
II. По возможному механизму развития.
1. С преимущественным вовлечением системы комплемента: наследственный и прибретенный.
1.1. Наследственный ангиоотек:
- 1-й тип – абсолютный дефицит С1-ингибитора (изолированный ангиоотек);
- 2-й тип – относительный дефицит С1-ингибитора (изолированный ангиоотек);
- 3-й тип – без дефицита С1-ингибитора (изолированный ангиоотек).
1.2. Приобретенный ангиоотек (приобретенный дефицит С1-ингибитора):
- 1-й тип – абсолютный (изолированный ангиоотек);
- 2-й тип – относительный c образованием аутоантител к С1-ингибитору (изолированный ангиоотек).
2. С участием других механизмов.
2.1. Вызываемый ингибиторами ангиотензин-превращающего фермента (АПФ) (изолированный ангиоотек).
2.2. Обусловленный гиперчувствительностью к лекарственным препаратам, пищевым продуктам, укусам и ужалениям насекомыми (в большинстве случаев сочетается с крапивницей).
2.3. Возникающий на фоне очаговой инфекции (может быть сочетанным).
2.4. Ассоциированный с аутоиммунными заболеваниями (может быть сочетанным).
3. Идиопатический (может быть сочетанным).
Из всего многообразия изолированных АО отдельного внимания заслуживают комплементзависимые ангиоотеки. Комплементзависимые отеки бывают наследственного и приобретенного характера. Для практической медицины наследственный АО (НАО) представляет серьезную проблему в связи с тяжестью обострений и высокой вероятностью развития тяжелых осложнений, которые в некоторых случаях приводят к летальному исходу.
Определение и классификация комплементзависимых ангиоотеков
Наследственный ангиоотек (НАО)
НАО – аутосомно-доминантное хроническое заболевание, относящееся к группе первичных иммунодефицитов, характеризующееся эпизодическим АО любой области тела. Причиной заболевания является снижение функции белка, ингибирующего С1-эстеразу, что приводит к неконтролируемой активации ряда белковых каскадов в сыворотке крови, главным образом классического пути активации комплемента и брадикининового каскада [1].
Наследственный комплементзависимый отек встречается редко и составляет не более 2% от всех случаев АО. В общей популяции распространенность НАО составляет 1:10 000 – 1:150 000 человек. Достоверных данных о распространенности НАО среди населения России нет в связи с низкой выявляемостью данного заболевания и отсутствием достаточной информированности врачей других специальностей об этом заболевании. НАО относят к разряду первичных иммунодефицитов. Больные НАО являются гетерозиготами, то есть имеют один нормальный и один измененный ген, отвечающий за синтез и функционирование С1-ингибитора. Мутация характеризуется аутосомно-доминантным типом наследования, хотя приблизительно 25% случаев НАО обусловлены спонтанной мутацией. Частота мутации гена составляет в среднем 1:100 000. В 2009 г. впервые среди населения РФ описан случай аутосомно-рецессивного наследования НАО, в зарубежной литературе описано лишь два случая подобного типа наследования. Несмотря на единичность этих данных, они существенно расширяют наши представления о типах наследования НАО [2].
Существуют два клинически идентичных типа НАО, определить которые можно лишь при исследовании компонентов комплемента в крови. Различают 2 фенотипических варианта заболевания: НАО 1-го типа и НАО 2-го типа. Некоторые исследователи выделяют 3-й тип НАО – эстрогензависимые отеки, однако до настоящего времени этот тип не выделен как самостоятельная нозологическая единица, так как не имеет четко и единообразно описанной этиологической и патогенетической картины. Отдельные авторы определяют его как наследственные эстрогензависимые ангиоотеки, то есть ангиоотеки, связанные исключительно с влиянием эстрогенов, с предположительным аутосомно-доминантным или Х-сцепленным доминантным типом наследования заболевания. Некоторые исследователи выделяют группу пациентов с рецидивирующими ангиоотеками и семейным анамнезом заболевания, у которых была выявлена локализованная в 5-й хромосоме мутация в гене, кодирующем 12-й фактор свертываемости крови. Наличие указанного генетического дефекта влечет за собой изменение его функциональной активности и, как следствие, нарушение кининового профиля, что приводит к повышению продукции брадикинина [3]. Отличительной особенностью болезни является зависимость симптомов от высокого уровня эстрогенов, соответственно, обострения болезни происходят во время беременности, применения пероральных контрацептивов или заместительной терапии эстрогенами при лечении климактерического синдрома. Первые две формы НАО обусловлены генетически детерминированным абсолютным или относительным дефицитом С1-ингибитора, при третьей его форме концентрация и функция С1-ингибитора не изменены.
НАО 1-го типа встречается у 85% всех пациентов с НАО. Болезнь отличается абсолютным дефицитом С1-ингибитора, при этом уровень С1-ингибитора варьирует в пределах 5–30% от нормальных значений. Дефицит С1-ингибитора обусловлен отсутствием гена, кодирующего С1-ингибитор, или различными его мутациями. Мутация заключается в разнообразных включениях, или делециях, одного или нескольких нуклеотидов в области гена, кодирующего С1-ингибитор. В случае если уровень С1-ингибитора нормальный или повышен, следует тестировать его функциональные характеристики для подтверждения НАО 2-го типа [1].
НАО 2-го типа составляет 15% среди всех типов НАО, отличается снижением функциональной активности С1-ингибитора, хотя уровень С1-ингибитора может оставаться в пределах или быть несколько выше нормы. У пациента вырабатывается нормальное или повышенное количество неправильно функционирующего С1-ингибитора. Нефункционирующий ингибитор не расходуется, накапливается в сыворотке крови и регистрируется в повышенном количестве.
Приобретенный ангиоотек (ПАО)
Помимо генетически детерминированного встречается приобретенный дефицит С1-ингибитора, что выражается приобретенными ангиоотеками (ПАО). ПАО отличается отсутствием у больных отягощенного наследственного анамнеза и поздним началом заболевания (в возрасте 40 лет и старше). Распространенные и редкие причины ПАО могут быть связаны с патологией желудочно-кишечного тракта (ЖКТ), гепатобилиарной системы и поджелудочной железы, системы органов дыхания, сердечно-сосудистой системы, нервной системы, опорно-двигательного аппарата, мочевыделительной системы, половых органов, эндокринного и метаболического генеза. ПАО может возникать при гематологических заболеваниях и иммунодефицитных состояниях, а также при распространенных инфекционных и паразитарных заболеваниях. Так, описаны случаи приобретенных комплементзависимых отеков на фоне ВИЧ-инфекции, множественной миеломы, различных лимфопролиферативных заболеваний, коллагенозов, хронических гепатитов В и С. Нередко приобретенные комплементзависимые отеки манифестируют за несколько лет до развития основного заболевания (табл. 1). Иногда такой АО является первым клиническим проявлением лейкоза [5].
Выделяют 2 типа приобретенных комплементзависимых отеков:
- ПАО 1-го типа связан с повышением катаболизма ингибитора С1 эстеразы при лимфопролиферативных заболеваниях;
- при ПАО 2-го типа происходит продукция IgG- и IgM-аутоантител к ингибитору С1 эстеразы, что приводит к его инактивации.
Развитие ПАО связано с образованием иммунных комплексов при лимфопролиферации, которые в избыточном количестве активируют систему комплемента, приводя к выработке большого количества анафилотоксинов.
Механизмы развития комплементзависимых отеков
Несмотря на прогресс в изучении биохимических маркеров и молекулярных характеристик наследственных отеков, механизмы, лежащие в основе запуска и разрешения атак, остаются неизвестными. Основной механизм отеков при НАО связан с эффектами медиаторов – брадикинина, гистамина, производных арахидоновой кислоты, цитокинов.
При наследственном дефекте системы комплемента нарушается продукция ингибитора С1 эстеразы вследствие мутации гена С1-ингибитора. Уровень С1-ингибитора снижен или он функционально малоактивен, что ограничивает активацию системы комплемента.
При дефиците С1-ингибитора увеличивается содержание калликреина, который, в свою очередь, повышает образование брадикинина. Недостаточность С1-ингибитора приводит к неконтролируемой активации ранних компонентов комплемента, локальный дефицит ингибитора С1 приводит к нарушению регуляции и повышению продукции вазоактивных пептидов. Накапливаясь в избыточном количестве, кинины вызывают расширение и повышение проницаемости сосудов микроциркуляторного русла, интерстициальный отек, набухание коллагеновых волокон, сглаживание сосочков дермы, что клинически проявляется отеком. В настоящее время обсуждается ведущая роль брадикинина в развитии симптомов комплементзависимых отеков. Брадикинин (БК) – это нанопептид, вырабатываемый при активации контактной системы, который может потенциально повышать сосудистую проницаемость путем связывания со своим рецептором (B2-рецептор к брадикинину) на эндотелиальных клетках сосудов. В зависимости от фармакологических свойств выделяют 2 подтипа рецепторов к брадикинину: BKR1 и BKR2 [7]. В настоящее время брадикинин считается ответственным за клинические симптомы ангиоотека, повышая сосудистую проницаемость, вызывая расширение сосудов и сокращение висцеральной гладкой мускулатуры. После воздействия инициирующего фактора недостаточность C1-ингибитора ведет к неадекватной выработке БК и повышению проницаемости сосудов, и выход жидкости в ткани вызывает незудящий отек.
При ПАО на фоне лимфопролиферативных заболеваний образуется большое количество антиидиотипических антител, формируются иммунные комплексы и происходит повышенное потребление С1q-компонента комплемента. В отличие от НАО, где синтез С1-ингибитора имеет дефект, ПАО характеризуется наличием большого числа аутоантител, действующих на молекулы C1q и, следовательно, на С1-ингибитор. Повышенный катаболизм С1q превосходит регуляторные возможности С1-ингибитора, создается относительный дефицит последнего, что приводит к развитию комплементзависимых отеков.
Клиническая картина комплементзависимых отеков
Для всех типов комплементзависимых отеков характерна особая клиническая картина, знание особенностей которой помогает в диагностике данной патологии.
АО провоцируют характерные особые триггеры. Любой из факторов, способных вызвать активацию комплемента по классическому пути, калликреин-кининовой системы или каскада свертывания крови, способствует возрастанию потребности в С1-ингибиторе и является потенциальным триггером обострения: механическая травма, интенсивность которой может быть разной – от легкого сдавления одеждой или легкого ушиба до перелома кости, – хирургическое вмешательство, малоинвазивные диагностические процедуры, экстракция зуба; лекарственные препараты: ингибиторы АПФ, пероральные контрацептивы, содержащие эстроген, препараты гормоно-заместительной терапии; инфекции, стресс, алкоголь [1, 6]. Хронические очаги инфекции, нередко не имеющие ярких клинических проявлений, такие как хеликобактерная инфекция, ассоциированы с более упорным течением НАО. Таким образом, острые приступы НАО возникают как спонтанно, так и под действием триггеров, причем 50% случаев НАО провоцируются травмой и хирургическими вмешательствами, а 30–40% приступов – психоэмоциональным стрессом. Молекулярные механизмы действия триггерных факторов на сегодняшний день изучены недостаточно. Исключение составляет применение ингибиторов АПФ у пациентов с НАО, которое в дальнейшем приводит к повышению уровня БК в тканях и плазме. Таким образом, не удивительно, что пациенты с НАО отвечают на прием ингибиторов АПФ увеличением частоты атак [7].
Для изолированных комплементзависимых АО характерна медленная динамика симптомов: отеки достаточно медленно нарастают в течение 12–36 часов и разрешаются в течение 2–5 дней, а абдоминальные симптомы исчезают в течение 12–24 часов. Пациенты указывают на неэффективность глюкокортикостероидов и антигистаминных препаратов при купировании отеков. Периодичность возникновения отеков может варьировать от еженедельных до 1 раза в год. Интервалы между обострениями у каждого больного индивидуальны: у нелеченых пациентов отеки могут возникать каждые 7–14 дней, также характерны ремиссии заболевания от 7–10 дней до 12 месяцев, возможны непрерывные атаки каждые 3 дня, а также латентное (субклиническое) течение. Данный значительный разброс в тяжести течения заболевания отмечается даже у родственников. Следует отметить, что у некоторых больных АО возникают только после значительной травмы, у других обострения проявляются достаточно часто, вне зависимости от внешних воздействий, на протяжении многих лет.
Атаки наследственных отеков обычно идут по предсказуемому пути. Многие атаки начинаются с продрома или ауры – обычно дрожь, покалывание, слабость, разбитость, мраморность кожи. Зуд и крапивница для НАО не характерны, однако примерно треть всех случаев АО сопровождается гигантской кольцевидной эритемой (erithema annulare), обильными бледными высыпаниями мультиформного характера, не сопровождающимися зудом, жжением и местной гиперемией.
Тяжесть симптомов наследственных отеков очень вариабельна и не коррелирует с плазменным уровнем С1-ингибитора [3, 6]. Другие факторы, такие как полиморфизм рецепторов к БК или вариации уровня или функциональности киназ, по всей вероятности, влияют на тяжесть болезни, но эти факторы не до конца изучены.
Симптомы ПАО развиваются после 4-го десятилетия жизни и нередко на несколько лет опережают другие симптомы субклинически протекающего лимфопролиферативного заболевания. В остальном клиника абсолютно идентична наследственным АО.
Диагностика комплементзависимых отеков
Существуют определенные трудности ранней диагностики НАО, особенно при атипичном течении заболевания. В связи с тем что несвоевременная постановка диагноза НАО обусловливает высокий процент смертности (20–30% пациентов умирают от отека гортани), всем пациентам с изолированными ангиоотеками следует проводить тщательное клиническое обследование, так как отечный синдром встречается не только при НАО, ПАО, но и вследствие других причин. Больным с отеками без симптомов крапивницы и кожного зуда следует уделять особое внимание, так как у них может быть НАО с синдромом недостаточности С1-ингибитора, носящий наследственный характер [5]. Очень важно проводить дифференциальную диагностику НАО с другими типами отеков (табл. 2).
При анализе анамнестических данных следует обратить внимание на семейный анамнез, возраст больного при дебюте заболевания, характерные триггеры, динамику развития отеков. Пациенты, страдающие рецидивирующим абдоминальным синдромом, при котором заболевания ЖКТ не подтверждаются лабораторно-функциональными и диагностическими методами, нуждаются в дополнительном обследовании для исключения патологии системы комплемента. Кроме того, следует обращать внимание на неэффективность традиционной терапии: антигистаминных препаратов, глюкокортикостероидов (эффект слабый или отсутствует), норадреналина, антибиотиков, противопаразитарных препаратов, ферментов. Лабораторное обследование является обязательным для подтверждения диагноза.
Согласно международным рекомендациям [9, 10], выделяют следующие клинические критерии диагноза НАО:
- Большие критерии:
- самостоятельно проходящий невоспалительный ангиоотек без уртикарной сыпи; медленно нарастающий и длящийся более 12 часов;
- спонтанно проходящая боль в животе неясной этиологии; часто рецидивирующая и длящаяся более 6 часов, может сопровождаться яркими диспептическими симптомами;
- рецидивирующий отек гортани.
- Малые критерии:
- семейный анамнез рецидивирующего ангиоотека, и/или болей в животе, и/или отека гортани;
- дебют заболевания в возрасте до 20 лет.
К лабораторным критериям диагноза НАО относят [9, 10]:
- уровни антигена C1-ингибитора эстеразы, составляющие
Читайте также: