
Синдром смита лемли опица реферат
Обновлено: 05.07.2024
ЧТО ЖЕ ТАКОЕ ПОЛИДАКТИЛИЯ?
ПРИЧИНЫ ПОЛИДАКТИЛИИ
ПРИЗНАКИ ЗАБОЛЕВАНИЯ
Основным признаком полидактилии является наличие у ребенка добавочных пальцев на кистях или стопах.
ТИПЫ ПОЛИДАКТИЛИИ
Врачи выделяют полидактилию трёх типов:
- Рудиментарная, при которой имеются добавочные пальцы, которые при этом состоят из кожи;
- Присутствие добавочных пальцев, которые являются раздвоением основных;
- Наличие полноценных, нормальной формы и размеров добавочных конечностей.
При этом полидактилия руки или ноги бывает:
- преаксиальной (радиальной) — со стороны большого пальца,
- центральной и постаксиальной — со стороны мизинца.
Важно отметить, что формы преаксиальной и постаксиальной полидактилии на практике встречаются гораздо чаще, чем центральная.
СИМПТОМАТИКА ЗАБОЛЕВАНИЯ
Полидактилия у человека наиболее часто проявляется, как удвоение мизинца на кисти руки либо как второй мизинец. Таким образом, на руке образуется не пять, а шесть пальцев. Часто случается, что дополнительный палец не до конца развит, в нем присутствуют лишь три фаланги, которые отходят от шестой пястной кости. Нередко встречаются даже одно- и двуфаланговый шестой палец. Также встречается такое проявление полидактилии, как кожный довесок, внутри которого вообще отсутствуют какие-либо костные образования. При такой патологии существенно нарушается моторная функциональность руки – такое возникает в случае развития дополнительного образования со стороны большого пальца.
СТАТИСТИКА
НАСЛЕДУЕТСЯ ЛИ ПОЛИДАКТИЛИЯ?
Да, патология очень часто передаётся именно по наследству, поскольку полидактилия – доминантный аутосомный признак. Вероятность унаследования патологии от отца или матери составляет около 50%. Ученые выделили 4 гена, от которых пальцы множатся.
Часто пациенты спрашивают: возможно ли предугадать полидактилию у ребенка? Ученые говорят, что довольно сложно предугадать, будет ли у ребенка полидактилия. Тип наследования или передача гена происходит по аутосомно-доминантному типу с определенным процентным соотношением. Но бывают и исключения, когда у шестипалых людей рождаются здоровые пятипалые дети.
ДИАГНОСТИКА ПОЛИДАКТИЛИИ
ПОЛИДАКТИЛИЯ, КАК ПРОЯВЛЕНИЕ РАЗНЫХ СИНДРОМОВ
Сама по себе изолированная полидактилия не несет дополнительных угроз организму, но важно понимать, что именно многопалость часто проявляется в составе сложных генных или хромосомных нарушений.
К примеру, полидактилия может быть признаком синдрома Патау или трисомии 13. Это значит, что в хромосомном наборе человека в 13 паре хромосом имеется одна лишняя хромосома (три вместо двух). И для этого заболевания характерно множественные нарушения развития внутренних органов и систем, в том числе, и полидактилия.
Синдром Рубинштейна — Тейби характеризуется аномалией развития пальцев, среди которых также встречается и полидактилия. Это связано с нарушением развития всей костной системы, костный возраст значительно отстает от паспортного, кости лицевого черепа деформированы, удваиваются либо концевые фаланги пальцев, либо весь палец.
Также полидактилия может быть признаком генетического заболевания, которое выражается в нарушении обменных процессов. Это синдром Смита-Лемли-Опица, но встречается он крайне редко.
Ученым на сегодня известно до 120 синдромов, признаком которых является шесть или даже больше пальцев на руках.
ОПРЕДЕЛЕНИЕ МЕТОДОВ ЛЕЧЕНИЯ
Для определения точных методов лечения лечащему врачу нужно установить:
— Характер патологии. При наследственной полидактилии добавочный палец отходит от пятого и развит довольно хорошо. Если речь о врожденном дефекте, то палец будет недоразвитым, в виде одной/нескольких фаланг внутри кожного выпячивания.
— Положение дополнительного пальца кисти, которое может быть преаксиальным (между безымянным пальцем и мизинцем) или постаксиальным — за мизинцем.
— Наличие дополнительных изменений в анатомии пясти или плюсны, которые могут проявляться в виде добавочных костей, деформаций суставов, связок и сухожилий.
— Является ли полидактилия самостоятельной аномалией или проявилась в составе синдрома. Для этого оцениваются другие симптомы и назначаются дополнительные анализы.
Только после этого выбирается точная методика лечения пациента. Подбирая методику оперативного лечения полидактилии, медики, кроме рентгенографии, проводят еще и исследование кровоснабжения добавочных пальцев, поскольку часто бывают случаи, когда крупный сосуд питает сразу два частично разделенных пальца. Тогда удаление без учета этой особенности может привести к последующим нарушениям кровоснабжения.
Когда все исследования завершены, проводится операция. Иногда ей предшествует терапевтическое лечение, но такие случаи единичны. Обычно лечащий врач настаивает на лечении полидактилии в раннем возрасте, поэтому большинство таких операций выполняется в первый месяц жизни ребенка.
ЛЕЧЕНИЕ
В случае подтверждения полидактилии, лечение может быть только хирургическое. Если лишний палец имеет только мягкие ткани, то его можно удалить в течение месяца после рождения младенца. В остальных случаях, когда лишний палец, кроме мягких тканей имеет кости, суставы и сухожилия, реконструктивная операция по ремоделированию кисти или стопы делается, как правило, через год после рождения ребенка.
При операции удаляется добавочный палец или кожный довесок. Необходимо провести операцию сразу после диагностирования, ведь патология может негативно отразиться на дальнейшем психологическом развитии ребенка или повлиять на мелкую моторику и функциональность конечностей.
Иногда может возникнуть вопрос, какой из пальцев лучше удалить – иногда диагностируют добавочный большой палец с хорошим развитием. Тогда удаляют тот, который расположен с внешней стороны кисти – так ей проще придать эстетичный вид.
Микрохирургическая коррекция полидактилии, в зависимости от её типа, может иметь различный характер, но почти всегда требуется костная, сухожильная и кожная пластика конечности.
ПРОГНОЗ
Исход операции практически всегда благоприятный, но очень важно сделать ее вовремя, тогда ребенок сможет нормально развивать моторику конечностей.
УНИКАЛЬНЫЕ СПОСОБНОСТИ ЛЮДЕЙ С ПОЛИДАКТИЛИЕЙ
ШЕСТИПАЛЫЕ ЗНАМЕНИТОСТИ

Мы заботимся о здоровье всех наших пациентов
Синдром Смита-Лемли-Опитца недавно стал объектом пристального внимания неврологов и психиатров из-за отчетливой взаимосвязи с аутистическими проявлениями. Возможно, данное заболевание имеет генетическую природу и тесно связано с аутистическими симптомами (см. ниже).
Синдром Смита-Лемли-Опитца (ССЛО) регистрируется с частотой около 1 на 70000 новорожденных. Характерна высокая перинатальная смертность и высокая частота мертворождений. Среди выживших детей не менее чем у 80% отмечаются тяжелые отклонения, а у оставшихся 20% изменения фенотипа выражены умеренно.
Синдром Смита-Лемли-Опитца (ССЛО) до сих пор диагностируется на основании типичных клинических признаков. Заболевание вызвано врожденным нарушением постскваленового синтеза холестерина. Нарушение синтеза холестерина при ССЛО вызвано наследственной мутацией гена 3-b-гидроксистерол-d-7 редуктазы (DHCR7). Повреждение гена DHCR7 приводит к нарушению синтеза холестерина и десмостерола, приводящее к повышению уровня 7DHC/8DHC, характерному снижению уровня холестерина и, что особенно важно, дисморфическому развитию.
Открытие синдрома Смита-Лемли-Опитца вызвало новые вопросы о роли путей биосинтеза холестерина в развитии человека. В настоящее время у 250 пациентов с синдромом Смита-Лемли-Опитца идентифицирована 121 мутация; разнообразие мутаций обеспечивает различную клиническую выраженность. Для воспроизведения некоторых аномалий развития, характерных для ССЛО, и выяснения патогенеза заболевания, было создано две генетических модели синдрома на мышах.
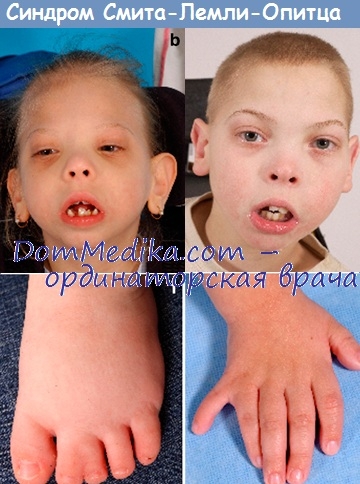
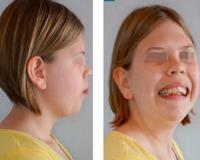
Наиболее характерными проявлениями синдрома наряду с задержкой умственного развития, аутизмом, другими поведенческими нарушениями и гипотонией является необычное строение лица с высоким плоским лбом, вывернутыми вперед ноздрями и микрогнатией, а также аномалии гениталий у мальчиков, включая крипторхизм и (или) гипоспадию, а в крайних случаях—неразвитые мужские гениталии, несмотря на нормальный кариотип XY (Scarbrough et al., 1986). Другие проявления включают аномальное строение ушных раковин, птоз век, эпикант, поперечное возвышение ладони и различные аномалии конечностей и внутренних органов (Opitz et al., 1994).
У некоторых пациентов отмечаются припадки, достаточно часто встречается отсутствие аппетита. В течение первого года жизни смертность высокая. Аномалии головного мозга могут включать микроцефалию, гипоплазию лобных долей, аномальное строение извилин и гипоплазию мозжечка. Случаи наиболее выраженного поражения иногда выделяются как ССЛО 2 типа (Curry et al., 1987).
Синдром наследуется аутосомно-рецессивным путем. Явное повышение частоты заболевания у мужчин, вероятно, связано с более легкой диагностикой на основании очевидных аномалий половых органов.
Концентрация холестерина в плазме очень низкая, в то время как содержание 7-гидрохолестерола повышено, что предполагает дефект фермента, приводящего к разрыву двойной связи гидрохолестерола (Tint et al., 1994). Выживаемость строго коррелирует с более высоким уровнем холестерина плазмы (Tint et al., 1995). Возможна пренатальная диагностика путем определения содержания 7-дегидрохолестерола в амниотической жидкости (Dallaire et al., 1995). Потребление большого количества холестерина может нормализовать его уровень в крови, но необязательно будет клинически эффективно (Ullrich et al., 1996).
Синдром Смита-Лемли-Опица — это редкое наследственное заболевание, которое связано с генетическим дефектом обмена холестерина. Возникает при мутации гена DHCR7 в 11-й хромосоме, в локусе 11q12-q13. Патология проявляется низкорослостью, умственной отсталостью, многочисленными костными аномалиями и врожденными пороками внутренних органов. Диагностика синдрома предполагает биохимические анализы, генетическое тестирование, инструментальные способы визуализации соответственно превалирующей симптоматике. Лечение поддерживающее: нейрореабилитационные мероприятия, помощь пластических хирургов, кардиохирургов, челюстно-лицевых хирургов.
МКБ-10
Общие сведения
Синдром назван в честь американских педиатров Д. Смита, Л. Лемли и генетика Дж. Опица, которые в 1964 г. описали его клинические признаки. Генетические механизмы развития заболевания были установлены в 1998 г. Частота встречаемости синдрома составляет в среднем 1:29000, чаще болеют европейцы (1:22000). При этом носительство мутантного гена широко распространено: до 1,2% у светлокожего населения и до 0,8% у темнокожих людей. Состояние характеризуется тяжелым течением, частой инвалидизацией, высокой смертностью, что обуславливает его актуальность в современной генетике.
Причины
Синдром Смита-Лемли-Опица связан с аномалией (миссенс- и нонсенс-мутацией) гена DHCR7, который располагается на длинном плече 11 хромосомы (11q12-q13). Он кодирует фермент 7-дегидрохолестерин-редуктазу, участвующий в заключительном этапе синтеза холестерина. Заболевание имеет аутосомно-рецессивный тип наследования. Риск возникновения синдрома у ребенка, оба родителя которого являются носителями поврежденного гена, составляет 25%.
Патогенез
Основным механизмом, который провоцирует появление патогномоничных фенотипических признаков синдрома, считается холестериновый дефицит. При этом заболевании конечная молекула не образуется, вместо нее в организме накапливаются липопротеины низкой плотности, другие промежуточные метаболиты. Эти вещества в избыточных количествах обладают токсическим влиянием на клетки.
Неврологические нарушения вызваны недостаточным образованием липидов, которые необходимы для миелинизации нервных волокон, а специфический сигнальный белок sonic hedgehog провоцирует нарушения внутриутробного формирования мозговой ткани. Дефицит холестерина коррелирует с недостатком гормонов (тестостерона, эстрогенов), что объясняет нарушения формирования наружных половых органов у мальчиков.
Симптомы
Для болезни Смита-Лемли-Опица специфичны микроцефалия, волчья пасть, микрогнатия. Реже встречается синдром Пьера-Робена в сочетании с выступающими резцами, что создает затруднения при жевании пищи, проведении анестезиологического пособия. У 95% детей наблюдается Y-образная синдактилия 2-3 пальцев ног, в половине случаев отмечается постаксиальная синдактилия. Также характерны вывих тазобедренных суставов, укорочение конечностей.
При генетическом дефекте DHCR7 типично отставание в росте, физическом развитии. С самого рождения младенцы плохо набирают вес, длина их тела ниже нормы. По мере взросления ребенка задержка роста становится более заметной, во взрослом возрасте параметры тела находятся ниже 3-го перцентиля. Нарушается половое созревание: у мальчиков выявляются гипоспадия, крипторхизм, у девочек — гипертрофия клитора.
У большинства пациентов определяется выраженная неврологическая симптоматика. Она проявляется нарушениями сна (70% больных): частыми ночными пробуждениями, неспособностью уснуть, отсутствием четкого режима дня. Психические симптомы включают повышенную агрессивность, в том числе аутоагрессию, склонность к навязчивому поведению, расстройства аутистического спектра. Большинство детей, страдающих синдромом Смита-Лемли-Опица, умственно отсталые.
Осложнения
36-38% случаев синдрома осложняется врожденными сердечными пороками. Аномалии желудочно-кишечного тракта встречаются у 25-29% больных. Они включают гастроэзофагеальный рефлюкс, болезнь Гиршпрунга, мальротацию кишечника. Распространены пороки мочеполовой системы: кистозная дисплазия почек (29%), дистопия почек (19%), гидронефроз (16%) и обструкция лоханочно-мочеточникового соединения (13%). Около 12-18% пациентов страдают от снижения зрения.
Большинству больных устанавливается инвалидность из-за выраженной умственной отсталости, отставания темпов физического развития. Синдром Смита-Лемли-Опица ассоциирован с высоким уровнем смертности как в перинатальном периоде, так и после рождения ребенка вследствие грубых врожденных аномалий. Хотя многие пациенты доживают до зрелого возраста, им требуется постоянный уход и медицинское наблюдение.
Диагностика
При физикальном осмотре ребенка удается выявить патогномоничные изменения фенотипа, что вместе с признаками задержки роста и психомоторного развития позволяет поставить предварительный диагноз. Для подтверждения синдрома Смита-Лемли-Опица назначается обследование у генетика, ряд диагностических мероприятий:
- Биохимический анализ. В плазме крови и тканях организма проверяется содержание липидов. При этом генетическом заболевании наблюдается повышение уровня 7-дегидорохолестерина одновременно со снижением количества холестерина.
- Генетическое исследование. Для обнаружения специфической мутации выполняется секвенирование экзона. При выявлении аномалии DHCR7 диагноз синдрома Смита-Лемли-Опица подтверждается со 100% точностью. Такая же диагностика показана родственникам больного.
- Пренатальная диагностика. Парам с отягощенной наследственностью рекомендуется исследование во время беременности путем измерения концентрации 7-дегидрохолестерола, генетического анализа амниотической жидкости или ворсин хориона.
- Инструментальные методы. Пациентам проводится комплексное обследование сердечно-сосудистой (эхокардиография, ЭКГ), мочеполовой (экскреторная урография, УЗИ органов малого таза), нервной системы (ЭЭГ, нейросонография, КТ или МРТ головного мозга).
Лечение синдрома Смита-Лемли-Опица
Консервативная терапия
При наличии синдрома Смита-Лемли-Опица проводится симптоматическая терапия, которая подбирается индивидуально на основании ведущих клинических признаков. Пациентам требуется помощь мультидисциплинарной команды врачей, поскольку у одного больного сочетаются разнообразные поражения внутренних органов, нарушения костно-мышечной системы, нервно-психические расстройства. Выделяют следующие направления лечения генетического синдрома:
- Диетотерапия. Для нормализации липидного профиля плазмы крови применяются пищевые добавки с кристаллическим холестерином, рацион обогащается яичными желтками, животными жирами.
- Реабилитация. Для улучшения моторных функций назначается комплексная программа ЛФК, массаж, кинезитерапия. Чтобы скорректировать нервно-психический дефицит, показаны длительные занятия с коррекционными педагогами, логопедами, психологами.
- Квалифицированный уход. Ввиду умственной отсталости пациенты не могут проживать самостоятельно. Уход за ними осуществляют родители с участием медицинского персонала, также рассматривается вариант пребывания больных в специализированных интернатах.
- Диспансерное наблюдение. Пациенты с синдромом Смита-Лемли-Опица должны регулярно проходить медицинские осмотры у ортопеда-травматолога, невролога, уролога и других специалистов. Частота обследований определяется тяжестью клинических симптомов.
Хирургическое лечение
Для устранения аномалий лицевого черепа, синдактилии, гипоспадии проводятся пластические и челюстно-лицевые вмешательства. Объем и сроки операции подбираются с учетом степени тяжести фенотипических изменений. Коррекция врожденных пороков сердца выполняется по стандартным протоколам в зависимости от характера аномалии, наличия расстройств кровообращения. Сложные пороки требуют вмешательства кардиохирургов уже на первом году жизни ребенка.
Прогноз и профилактика
При своевременной диагностике синдрома и раннем начале комплексной терапии прогноз относительно благоприятный. Врачам удается ускорить психомоторное развитие, устранить аномалии строения скелета, скорректировать соматические патологии. Однако низкий рост, умственная отсталость, проблемы с фертильностью остаются на всю жизнь. Профилактика предполагает генетическое консультирование носителей мутировавшего гена перед планированием беременности.
1. Федеральные клинические рекомендации по диагностике и лечению синдрома Смита-Лемли-Опица/ Л.П. Назаренко. — 2015.
2. Врожденные пороки развития. Синдром Смита-Лемли-Опица/ О.А. Милованова, Н.В. Чернышева, А.И. Чубарова, Л.В. Калинина// Неврологический журнал. — 2013. — №3.
3. Случай синдрома Смита-Лемли-Опица/ Л.И. Ширяева, А.М. Поздняков, Е.А. Соляник// Научно-медицинский вестник Центрального Черноземья. — 2006. — №24.
4. Синдром Смита-Лемли-Опица у детей/ А.Н. Семячкина, П.В. Новиков, М.И. Яблонская, М.Б. Кубатов// Российский вестник перинатологии и педиатрии. — 2006. — №3.
Синдром Смита-Лемли-Опица (SLO) представляет собой нарушение обмена веществ, которое включает в себя несколько различных симптомов, таких как значительно медленный рост, характерные легкие признаки, микроцефалия (измерение головы меньше нормального), умственная отсталость, которая может быть легкой или умеренной, трудности в обучении и проблемы поведение.
Это также сопровождается пороками развития легких, сердца, почек, кишечника и даже половых органов. Кроме того, они могут представлять синдактилию или слияние некоторых пальцев или полидактилию; это означает, что у них более 5 пальцев на ноге или руке.

По-видимому, это связано с отсутствием фермента, который важен для метаболизма холестерина, который приобретается при генетическом наследовании аутосомно-рецессивного паттерна..
Тем не менее, такие проявления, кажется, сильно различаются в зависимости от тяжести заболевания даже в одной семье.
Этот синдром может встречаться в литературе под такими названиями, как: дефицит 7-дегидрохолестерин редуктазы, синдром RSH или синдром SLO.
Немного истории .
В 1964 году педиатры Дэвид Смит, Люк Лемли и Опитц Джон описали 3 пациентов мужского пола с микроцефалией и гипогенитализмом и определили это состояние как RSH по инициалам первоначальных фамилий этих пациентов..
Впоследствии название синдрома было изменено на фамилии первооткрывателей (SLO).
Примерно через 30 лет Tint et al. (1994) обнаружили у 5 пациентов с этим состоянием значительно более низкие концентрации холестерина в крови, но увеличение более чем в 1000 раз уровня 7-дегидрохолестерина. Они увидели, что это увеличение связано с отсутствием фермента, который должен преобразовывать 7-дегидрохолестерин в холестерин.
Позже был идентифицирован ген DHCR7, связанный с этим заболеванием, клонирование в 1998 году (Witsch-Baumgartner & Lanthaler, 2015).
статистика
Синдром Смита-Лемли-Опитца затрагивает приблизительно 1 на 20 000–60 000 живорождений во всем мире. Он действительно может быть унаследован от 1 из 1590 до 13 500 особей, но эта цифра не используется, потому что многие плоды с этим заболеванием умирают до своего рождения (Национальная организация по редким заболеваниям, 2016).
Что касается секса, то он одинаково влияет как на мужчин, так и на женщин, хотя его легче диагностировать у мужчин, поскольку пороки развития половых органов более заметны, чем у женщин. Кроме того, это, кажется, чаще встречается у людей европейского происхождения; особенно из стран, принадлежащих к центральной Европе, таких как Чехия или Словакия. Тем не менее, это очень редко в популяции Африки или Азии.
Причины синдрома Смита-Лемли-Опица
Синдром SLO появляется из-за мутаций в гене DHCR7, присутствующем в хромосоме 11, который отвечает за отправку заказов на производство фермента 7-дегидрохолестерин редуктазы. Это фермент, который модулирует выработку холестерина и будет отсутствовать или очень мало при этом синдроме, что приводит к недостаточной выработке холестерина, что будет препятствовать нормальному росту.
Это оказывает большое влияние, так как холестерин важен для организма. Он состоит из липида, похожего на жир, который получают в основном из продуктов животного происхождения, таких как яичные желтки, молочные продукты, мясо, птица и рыба.
Важно, чтобы эмбрион развивался без затруднений, выполняя важные функции, такие как вклад в структуру мембран клеток и миелина (вещества, которое покрывает клетки мозга). Он также служит для производства гормонов и пищеварительных кислот.
Недостаток фермента 7-дегидрохолестеринредуктазы вызывает накопление в организме компонентов, которые могут быть токсичными для холестерина. Таким образом, мы имеем, с одной стороны, низкий уровень холестерина и в то же время накопление веществ, которые могут быть токсичными для организма; вызывая отсутствие роста, умственную отсталость, физические уродства и проблемы во внутренних органах.
Однако с полной уверенностью не известно, как эти проблемы, связанные с холестерином, приводят к симптоматике синдрома Смита-Лемли-Опитца..
В настоящее время в гене DHCR7 обнаружено более 130 мутаций, связанных с синдромом, фактически существует база данных, которая включает все описанные случаи SLO с ее вариантами, фенотипами и генотипами..
Хотя существует так много возможных мутаций, большинство случаев относится к 5 наиболее частым, а остальные очень редки (Witsch-Baumgartner & Lanthaler, 2015).
Эти мутации в гене DHCR7 наследуются по аутосомно-рецессивному типу, это означает, что человек, представивший синдром, должен был унаследовать мутированный ген от двух родителей. Если вы получите его только от одного из родителей, вы не представите болезнь; но это может быть носитель и передать его в будущем.
Существует 25% риска того, что у двух родителей-носителей есть больной ребенок, в то время как риск того, что ребенок будет носителем, также будет составлять 50% при каждой беременности. С другой стороны, в 25% случаев он может родиться без этих генетических мутаций или быть носителем; все эти данные не зависят от пола ребенка (Национальная организация по редким заболеваниям, 2016 г.).
Имейте в виду, что вероятность рождения детей с генетическим рецессивным расстройством выше, если родители являются близкими родственниками (или родственниками), чем родители, у которых нет таких связей.
Какие симптомы это имеет??
Симптомы варьируются в зависимости от пострадавшего человека, в зависимости от количества холестерина, которое они могут производить.
По словам Хименеса Рамиреса и соавт. (2001), клинические характеристики охватывают несколько аспектов и могут быть очень разнообразными. Как правило, они находятся на лице, конечностях и половых органах; хотя они могут включать другие телесные системы.
Многие из пострадавших имеют типичные признаки аутизма, влияющие на социальное взаимодействие. Если состояние мягкое, можно увидеть только некоторые проблемы в обучении и поведении; но в наиболее серьезных случаях у человека могут быть серьезные нарушения интеллекта и физические отклонения, которые могут привести к смерти.
Есть симптомы, которые могут уже присутствовать с рождения человека, хотя мы будем включать те, которые встречаются на всех этапах жизни:
У более чем 50% пациентов:
- Недостаток физического развития наблюдается после рождения.
- Умственная отсталость (100%).
- Микроцефалия (90%).
- Синдактилия или слияние 2 или 3 пальцев (

Многие причины врожденных аномалий известны, около 50 % случаев остаются невыясненными. Хромосомные нарушения составляют около трети всех наблюдаемых врожденных аномалий. Как правило, хромосомный дисбаланс ведет к множественным тяжелым врожденным порокам развития, иногда несовместимым с жизнью. Моногенные дефекты составляют около 7,5 % всех врожденных аномалий. Некоторые из них ведут к изолированным врожденным аномалиям, другие – к синдромам множественных врожденных пороков, затрагивающих многие органы и их системы. Например, эктродактилия может наследоваться как аутосомно-доминантный, аутосомно-рецессивный и, редко, как
Х-сцепленный признак. Она также может входить в симптомокомлекс определенного синдрома (например, аутосомно-доминантный синдром EEC – экодермальная дисплазия, эктродактилия и расщелина губы и неба).
Мультифакторное наследование характерно для ряда несиндромальных врожденных аномалий сердца, центральной нервной системы. Для многих из них известны риски, выявленные эмпирическим путем на основе эпидемиологических исследований. Для ряда специфических врожденных мальформаций характерна генетическая гетерогенность. Некоторые наиболее частые врожденные аномалии представлены ниже.
Голопрозенцефалия. Эта тяжелая и часто летальная мальформация представляет собой нарушение развития такой области эмбрионального мозга, как передний мозг или прозенцефалон. Голопрозенцефалия характеризуется полным или частичным нарушением деления переднего мозга по средней линии и формирования гемисфер, что ведет к нарушениям не только структуры мозга, но и к развитию срединных лицевых структур (например, возникновению циклопии), а также к глубоким нарушениям психомоторного развития. Тяжесть аномалий лица обычно соответствует тяжести поражения структур мозга. Частота голопрозенцефалии составляет около 6–12 на 10000 среди новорожденных. Половина этих случаев связана с трисомией хромосомы 13. При голопрозенцефалии наблюдается преобладание лиц женского пола.
Голопрозенцефалия может быть этиологически классифицирована на хромосомную, синдромальную и изолированную. Причины, связанные с аномалиями хромосом, составляют 30–40 % случаев голопрозенцефалии, наиболее частой является трисомия хромосомы 13. Среди других причин могут быть делеции хромосом 2p11, 7q36, 18р11.3 и 21q22.3, дупликации хромосом 3р24→pter и 13q, а также триплоидии. Синдромальные причины голопрозенцефалии многочисленны, например синдром Смита-Лемли-Опица. Причинами изолированной голопрозенцефалии могут быть мутации ряда генов (SHH, ZIC2, SIX3).
Дефекты нервной трубки. Эти врожденные пороки являются результатом аномального закрытия развивающейся нервной трубки в эмбриональном периоде. Дефект верхнего конца развивающейся нервной трубки ведет либо к анэнцефалии (врожденное отсутствие головного или спинного мозга), либо к энцефалоцеле (выпячивания мозга через имеющийся в черепе дефект). Дефекты нижнего конца нервной трубки ведут к таким спинальным порокам, как пояснично-крестцовое миелоцеле или менингомиелоцеле (выпячивания спинного мозга и его оболочек через незаращение дужек позвонков). Анэнцефалия несовместима с жизнью. Крупные пороки в пояснично-крестцовой области вызывают частичный или полный паралич нижних конечностей с нарушением функций мочевого пузыря и кишечника.
Дефекты нервной трубки также могут быть этиологически классифицированы на хромосомные, синдромальные и изолированные. Причины, связанные с аномалиями хромосом, включают трисомии хромосом 13 и 18, при которых дефекты нервной трубки встречаются в 5–10 % случаев. Среди моногенных причин следует отметить относительно редкое аутосомно-рецессивное заболевание – синдром Меккеля, при котором энцефалоцеле сочетается с поликистозом почек и полидактилией. Тем не менее, большинство дефектов нервной трубки представляют собой изолированные пороки и наследуются мультифакторно. Эмпирический риск варьирует согласно региональной частоте и достигает 4–5 %. Например, у жителей Англии частота данной патологии выше, чем у остальных национальностей кельтского происхождения.
Известно, что мутации в гене метилентетрагидрофолатредуктазы являются предрасполагающим фактором к дефектам нервной трубки. Мутация данного гена ведет к снижению активности одноименного фермента, являющегося ключевым в обмене фолиевой кислоты, и наиболее опасна в условиях дефицита фолатов в питании. Нарушения обмена фолатов ведут к изменениям образования пуринов и пиримидинов, необходимых для синтеза ДНК. Профилактика, проводимая при помощи введения в диету витаминов, в частности, фолиевой кислоты за несколько месяцев до зачатия, а также в первые месяцы беременности уменьшает риск повторного рождения ребенка с пороком нервной трубки в семье на 70–75 %. Во многих странах (США, Великобритания, Венгрия, Мексика) принято обогащать муку и хлеб фолиевой кислотой, что приводит к заметному снижению частоты пороков нервной трубки.
Читайте также: