
Синдром корнелии де ланге реферат
Обновлено: 03.07.2024
Синдром Корнелии де Ланге (синдром Брахмана де Ланге, синдром дегенеративного нанизма амстердамского типа) представляет собой редкое заболевание, впервые описанное Брахманом в 1916 г., но носящее имя датского педиатра де Ланге. Синдром считается очень редким, частота его составляет всего один случай на 100000 новорожденных (Beck, 1976).
а) Патогенез. Синдром Корнелии де Ланге сопровождается мутациями гена NIBPL короткого плеча пятой хромосомы (Gillis et al., 2004; Krantz et al., 2004; Tonkin et al., 2004). Недавно было выявлено, что описанные мутации является основным этиологическим фактором данного синдрома и выявляются у 27-56% пациентов (Yan et al., 2006).
Тем не менее, среди пациентов с синдромом Корнелии де Ланге выявляется ряд других хромосомных аномалий (Jackson et al., 1993). Проявления, позволяющие выявить наличие данного синдрома, отмечаются при частичной трисомии дистальной части 3-й хромосомы (3q21-3ter) и других перестройках 3-й хромосомы (DeScipio et al., 2005). Синдром дупликации 3q хромосомы (dup (3q)-синдром) только на первый взгляд имитирует синдром де Ланге (Holder et al., 1994).
Данные изменения и изменения, связывающие ген МЕСР2 с синдромом Ретта, доказывают, что следует соблюдать осторожность, рассматривая ген NIBPL как единственную причину развития синдрома де Ланге.
Синдром де Ланге может наследоваться доминантным путем; в действительности все случаи синдрома представляют собой вновь возникшие мутации. Риск повторного рождения ребенка с данной патологией составляет 2-5%.
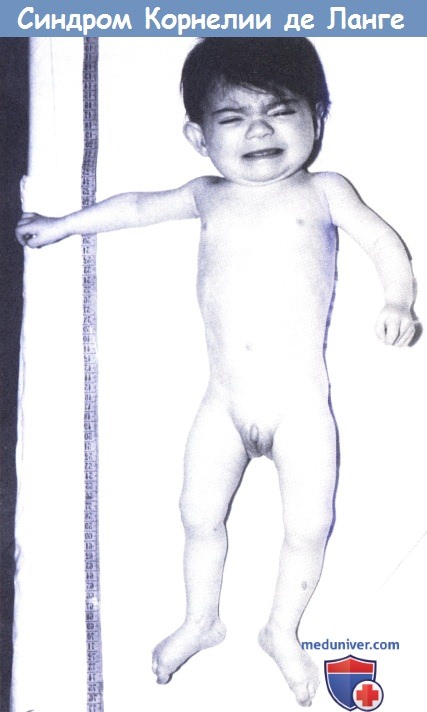
Синдром Корнелии де Ланге:
вдавленная переносица, низкая линия роста волос, сросшиеся брови,
короткие руки и согнутые пальцы рук и ног.
б) Клинические проявления. Синдром де Ланге является мультисистемным заболеванием, проявляющимся пре- и постнатальной задержкой роста, замедленным развитием, характерным дисморфизмом лица, мальформациями конечностей и множественными поражениями органов.
Кроме того, данное заболевание проявляется достаточно характерным поведенческим фенотипом, и в большинстве случаев серьезной или полной необучаемостью (Horsier и Oliver, 2006), часто сочетающейся с самодеструктивным поведением и избеганием социальных контактов, нередко достигая степени синдрома аутизма (Gillberg и Coleman, 2000; Arron et al., 2006; Bhuiyan et al., 2006).
Среди пациентов с мутациями отмечается тенденция к более выраженным изменениям массы тела, роста и средней окружности головы при рождении, дисморфизму лица и нарушению речи, чем среди пациентов, у которых мутации отсутствуют.
Клинически очерченные генетические синдромы с неясным ходом наследования представляют собой отдельную группу заболеваний, характеризующихся своеобразным сочетанием различных множественных врожденных аномалий, которые придают соответствующий облик каждому больному, и довольно глубокой умственной отсталостью. Этиология их остается неясной. За генетический характер этих синдромов говорят следующие данные: семейное накопление макро- и микропризнаков, встречающиеся случаи кровного родства родителей.
Содержание
ВВЕДЕНИЕ 2
1. Синдром Карнелии де Ланге 4
2. Клиническая картина 5
ЗАКЛЮЧЕНИЕ 10
Список литературы 10
Прикрепленные файлы: 1 файл
синдром де ланге.docx
1. Синдром Карнелии де Ланге 4
2. Клиническая картина 5
Список литературы 10
ВВЕДЕНИЕ
Клинически очерченные генетические синдромы с неясным ходом наследования представляют собой отдельную группу заболеваний, характеризующихся своеобразным сочетанием различных множественных врожденных аномалий, которые придают соответствующий облик каждому больному, и довольно глубокой умственной отсталостью. Этиология их остается неясной. За генетический характер этих синдромов говорят следующие данные: семейное накопление макро- и микропризнаков, встречающиеся случаи кровного родства родителей. Некоторые авторы предполагают полигенный характер наследования этих заболеваний, другие считают, что они обусловлены вновь возникшими доминантными мутациями; предполагается также и специфическая наследственная предрасположенность в сочетании с рядом средовых факторов, и вероятность невыявленных хромосомных мутаций.
Эту группу заболеваний составляет ряд синдромов, хотя и относимых к генетически обусловленным, но встречающихся главным образом спорадически. Однако описания монозиготных близнецов с данной патологией всегда свидетельствуют об их конкордантности; кроме того, иногда встречаются семейные случаи этих заболеваний.
Наиболее часто встречающиеся синдромы этой группы наследования: синдром Рубинштейна — Тейби, умственная отсталость с гипертрихозом, синдром Корнелии де Ланге (амстердамская карликовость). В данной работе мы хотим изучить клиническую картину синдрома Корнелии де Ланге,
Задачи данной контрольной работы:
- Описать клиническую картину при синдроме Корнелии де Ланге
- Изучить особенности проявления физических нарушений, умственной отсталости при данном синдроме.
- Рассмотреть специфику диагностики и возможности лечения синдрома.
Синдром Карнелии де Ланге
Синдром Де Ланге (Брахмана – Ланге синдром, описан немецким врачом W. R. C. Brachmann, род. в 1888, позднее – нидерландским педиатром С. de Lange, 1871–1950) – врожденное заболевание наследственной природы, проявляющееся умственной отсталостью и множественными аномалиями развития. Встречается довольно редко, но практически все случаи сопровождаются различными хромосомными нарушениями.
Предполагают, что синдром де Ланге может быть обусловлен разными генетическими механизмами. Тип наследования не уточнен. Подавляющее большинство случаев заболевания — спорадические. Вместе с тем известны семейные случаи, на основании анализа которых высказывается мнение об аутосомно-рецессивном типе наследования. Однако семейные заболевания обычно отличаются от спорадических отсутствием редукционных пороков развития конечностей, что позволяет предполагать различную этиологию семейных и спорадических случаев. Данные о влиянии возраста отцов на частоту возникновения синдрома противоречивы, в связи с чем остается неясным, могут ли спорадические аутосомно-доминантные мутации быть его причиной. У подавляющего большинства больных кариотип нормальный, однако, в ряде случаев обнаружены его аномалии: из которых наиболее частыми являются частичная трисомия длинных плеч хромосом 1 и 3, хромосома 9 в виде кольца.
2. Клиническая картина синдрома Корнелии де Ланге
Характерный признак болезни – специфическое строение лица: нос очень короткий с повернутыми вперед ноздрями и запавшей переносицей, расстояние между носом и верхней губой значительно больше нормы, губы тонкие - их уголки опущены вниз, словно от обиды. Помимо этого, дети с синдромом Корнелии де Ланге отличаются весьма густыми, нередко сросшимися бровями, а также длинными ресницами, по-кукольному загнутыми вверх. Голова у больных малышей, как правило, уменьшена в размерах. Наблюдается отставание в росте и весе.
И все же диагностируют синдром Де Ланге по другому признаку, его считают основным - это неправильное строение пальцев на руке. Малыши рождаются либо с искривленным мизинчиком и укороченными указательными пальчиками, либо большие пальцы рук почти прижаты к ладони. Часто встречаются деформации суставов, грудины, позвоночника. Описаны случаи различных аномалий почек. Дети с данным синдромом могут страдать сильным оволосением в области поясницы и вдоль позвоночника.
Для диагностики синдрома важное значение имеет оценка костносуставных деформаций. Частыми являются контрактуры или тугоподвижность в локтевых суставах, искривление мизинца, укорочение второго и отведение первого пальцев рук. Могут наблюдаться также более тяжелые дефекты в строении конечностей с недоразвитием их отдельных сегментов. В некоторых случаях встречаются деформации позвоночника и грудины. Среди пороков внутренних органов преобладают аномалии строения почек.
Как уже отмечалось, характерным признаком синдрома считается усиленный рост волос, особенно выраженный на спине и в поясничной области, а также на наружной поверхности предплечий. Часто наблюдается также мраморность кожи, краснота кончика носа и цианоз носогубной области.
Дети с данным заболеванием обычно рождаются с малым весом и ростом.
При рождении малыша с характерными признаками синдрома Де Ланге, его сразу показывают неврологу, ортопеду, а затем педиатру. Эти врачи и будут в дальнейшем наблюдать ребенка, поскольку с возрастом у деток возникают сложности с умственным отставанием и различными нарушениями поведения. Дети с трудом осваивают речь, им сложно общаться с окружающими, они способны к самоагрессии и отрицанию норм социального поведения.
В первые годы жизни дети отличаются соматической ослабленностью, повышенной восприимчивостью к простудным и инфекционным заболеваниям верхних дыхательных путей; часто болеют бронхитами, пневмониями. С первых месяцев жизни они отстают в физическом и психомоторном развитии.
Диагноз устанавливают на основании характерного фенотипа. Наиболее постоянными признаками являются микробрахицефалия, синофрис, особенности строения носа и увеличенное расстояние между его основанием и верхней губой, микромелия, гипертрихоз, отставание в физическом и нервно-психическом развитии. Обязательно исследование кариотипа с использованием современных методов цитогенетического анализа.
На коже у больных, кроме гипертрихоза, резко выраженного в области спины и поясницы, нередко отмечается общая мраморность, характерны краснота кончика носа, цианоз носогубной области.
Среди неврологических нарушений, как правило, наблюдаются мышечная гипотония, оживление сухожильных рефлексов, но возможен и гипертонус с мышц конечностей.
Диагноз синдрома Корнелии де Ланге не всегда прост, так как встречаются умственно отсталые дети с небольшим числом аномалий, входящих в данный синдром. При отсутствии несомненного биологического метода диагностики трудно сказать, можно ли такие случаи относить к синдрому Корнелии де Ланге. Как известно, этот сложный вопрос касается и других заболеваний, диагностика которых осуществляется на основе только клинического своеобразия.
Умственная отсталость определяется практически у всех больных с данным синдромом: в 80 % случаев устанавливают имбецильность или глубокую дебильность. Однако описаны и больные с нерезко выраженным интеллектуальным дефектом (IQ = 73-75).
Для заболевания характерны системное недоразвитие речи, нерезко выраженные формы дизартрии. Иногда может наблюдаться длительная тенденция говорить о себе в 3-м лице.
У детей с данным синдромом часто наблюдаются нарушения поведения в виде повышенной аффективной возбудимости, двигательной расторможенности, склонности к истероформным реакциям. В литературе есть указания на то, что у больных с данным синдромом имеются стремление к аутоагрессии и склонность к стереотипным движениям - бегу по кругу, вращению, стереотипным движениям руками. Судорожный синдром отмечается у 25 % больных: судороги, как правило, только эпизодические, а не в виде частых и полиморфных приступов. В силу тяжести и осложненного характера интеллектуального дефекта обучаемость детей обычно низкая, но вместе с тем рано начатые с ними кор-рекционные занятия оказывают положительное влияние на дальнейшее психическое развитие ребенка.
При рентгенографии черепа нередко обнаруживаются явления внутричерепной гипертензии. При электроэнцефалографии каких-либо специфических, характерных для синдрома изменений не выявляется.
Патологическая анатомия при синдроме Корнелии де Ланге изучена достаточно подробно. Как наиболее характерное нарушение мозга описывается двусторонняя аплазия оперкулярных отделов лобных долей. Отмечают также отсутствие центральной (роландовой) борозды, гипоплазию пирамид обонятельного нерва, верхних височных извилин, задней спайки мозолистого тела, отставание миелинизации и распад миелина, глиоз и очаговую аплазию клеток наружного зернистого и пирамидного слоев во всех отделах мозга.
Этиология синдрома остается неясной. Несомненно, поражение возникает в ранние сроки эмбрионального развития. Описано значительное число случаев синдрома Корнелии де Ланге с различными хромосомными нарушениями, однако роль их неясна. Наряду с этим наблюдались семьи с пораженными сибсами (без хромосомной патологии), что позволяет некоторым авторам высказывать мнение об аутосомно-рецессивном наследовании синдрома. Известны также случаи, когда заболевание передается от родителей, имеющих неглубокий дефект и соматические черты синдрома.
Лечение. Специфического лечения не существует. Применяют ноотропные средства, анаболические гормоны, витамины.
При необходимости проводят противосудорожную и седативную терапию. Применяют ноотропы, анаболические гормоны (неробол, ретаболил), назначают витаминотерапию. Прогноз для жизни неблагоприятный, большинство больных погибают в раннем детстве.
Профилактика заключается в медико-генетическом консультировании (семей, в которых имеются больные. Эмпирический риск повторного рождения ребенка с синдромом де Ланге в спорадических случаях при отсутствии хромосомных аберраций у родителей составляет 2%. В семейных случаях, исходя из гипотезы аутосомно-рецессивного наследования синдрома, вероятность рождения больного ребенка составляет 25%.
Для побуждения умственной активности таким детям будут полезны музыкальные занятия: игры с музыкальным сопровождением, ритмичные движения, спокойное слушание напевных мелодий, показ и участие в сказочных представлениях. Особое внимание взрослые должны уделять развитию речи. Сегодня разработано немало специальных программ по обучению деток с Де Ланге и другими похожими синдромами. Простые потешки, считалки, стихи важно разучивать с малышом терпеливо, целенаправленно, используя наглядные пособия и игры. Случается, что дети совсем не поддаются обучению речи - обратитесь к врачу-сурдологу, возможно у малыша проблемы со слухом (распространенные случаи).
Статья посвящена редкому синдрому с неясным ходом наследования. Описываются этиология, фенотипические признаки, симптомы, на основании которых педиатр и генетик могут заподозрить амстердамскую карликовость. Кратко изложены общие принципы лечения. Приведено клиническое наблюдение у ребенка Р. в возрасте 16 лет с полиорганным поражением.
Ключевые слова
Для цитирования:
For citation:
Введение
Синдром Корнелии де Ланге - это редкая врожденная патология (относится к орфанным болезням), характеризующаяся тем, что ребенок рождается со сразу заметными множественными стигмами дизэмбриогенеза. Впоследствии у младенца обнаруживаются еще и признаки умственной отсталости [1][2].
Заболевание встречается у новорожденных с частотой от 1:30000 до 1:10000, соотношение мальчиков и девочек — 1:1. Тип наследования не уточнен, известны семейные случаи с аутосомно-рецессивным типом наследования. У большинства больных кариотип нормальный. Синдром является генетически гетерогенным, обусловленным мутациями в гене NIPBL, SVC3, микродупликацией локусов q25-q29 хромосомы 3. Всего на данный момент известно более 400 случаев этого заболевания в разных странах [3][4].
В последнее время предполагают влияние на развитие данной патологии ряда следующих факторов риска:
- Наличие в семейном анамнезе этого синдрома, т.к. в этом случае (если предположение о рецессивном способе передачи гена верно) возможность появления следующего ребенка с патологией составляет 25%.
- Степень вероятности повторения ситуации в одиночных эпизодах при отсутствии хромосомных мутаций у родителей теоретически равна 2 %.
- Преобразования хромосом могут возникать вследствие тяжелых инфекций и интоксикаций, перенесенных будущей матерью в первые три месяца беременности, побочных действий химиотерапевтических лекарственных средств и некоторых физиотерапевтических процедур.
- Генным мутациям могут способствовать эндокринные заболевания матери и радиация.
- Солидный возраст отца ребенка, либо материнский возраст более 35 лет.
- Мать и отец — кровные родственники.
В клинической картине синдрома выделяют следующие основные признаки:
Первые признаки заболевания визуально заметны уже у новорожденных. Кроме внешних особенностей, обращает на себя внимание маленький вес ребенка при рождении, который составляет 2/3 веса здорового ребенка, родившегося на аналогичном сроке беременности. Для новорожденных характерно наличие респираторного дистрегс-синдрома из-за специфического строения носоглотки, поэтому в анамнезе у этих пациентов регистрируются частые инфекционно-воспалительные заболевания дыхательных путей [6][7][8].
При вскрытии умерших больных обнаруживаются разнообразные дефекты головного мозга (недоразвитие нижней лобной извилины, расширение желудочков, дисплазия и гипоплазия извилин), гистология нередко показывает выраженную поперечную исчерченность нейронов внешнего зернистого слоя коры больших полушарий и расстройство топографии нейронов мозжечка.
Более чем в половине всех случаев амстердамскому нанизму сопутствуют дефекты в структуре сердца (аортолегочное окно; незаращенная перегородка, разделяющая как предсердия, так и желудочки, часто в комбинации с сосудистыми нарушениями; тетрада Фалло), дефекты в структуре ЖКТ (в основном, нарушения поворота кишечника), мочеполовой системы (кистозные образования почек, одиночные и множественные, иногда подковообразная почка и гидронефротические ее изменения, крипторхизм, двурогая матка) [9][10].
Это заболевание, характеризующееся множеством дефектов развития, является по своей сути пока еще не раскрытой генетической аномалией, которая начинается в период формирования эмбриона. Процесс, запущенный патогенным фактором, продолжается и усугубляется в дальнейшем, после рождения ребенка.
Выделяют два варианта синдрома: классический, со значительной задержкой физического и интеллектуального развития, грубыми пороками развития, и стертый, с лицевыми и малыми скелетными аномалиями, но пограничной задержкой психомоторного развития и отсутствием грубых пороков. Диагноз устанавливают на основании фенотипа, исследования кариотипа и методов цитогенетического анализа [2][6].
Специфического лечения не существует. Применяют нейрометаболические, ноотропные препараты, витамины, симптоматическую терапию, коррекцию логопедическую, психологическую.
Профилактика
Профилактикой синдрома, факторы возникновения которого точно не установлены заниматься сложно. Однако с учетом известных источников генных мутаций можно рекомендовать в качестве профилактических мер:
- предотвращение зачатия детей от матери и отца, являющихся кровными родственниками;
- тщательно обследоваться в случае возможности позднего материнства и отцовства;
- беременным женщинам следует избегать заражения вирусными инфекциями, особенно в первом триместре, а в случае заражения, применять лекарственную терапию только по назначению врача;
- женщины и мужчины, в семейном анамнезе которых были случаи синдрома Корнелии де Ланге, обязательно должны посетить медико-генетическую консультацию;
- во время беременности женщинам, в семейном анамнезе которых наблюдались случаи синдрома Корнелии де Ланге, обязательно нужно обследоваться на наличие протеина-А плазмы крови.
Прогноз
Продолжительность жизни зависит от многих факторов, главными из них можно назвать степень тяжести пороков жизненно важных органов, их раннюю диагностику и качество хирургических вмешательств по их ликвидации.
При аномалиях развития, несовместимых с жизнью, ребенок умирает на первой неделе жизни. В случае их незначительности или своевременного устранения хирургическим путем, больной с синдромом Корнелии де Ланге может прожить достаточно долго. Прогнозирование осложняется отсутствием сопротивляемости организма больных с данным синдромом ординарным, неопасным для обычных людей инфекциям, например, вирусным, которые тоже становятся причиной ранней смерти таких больных.
Средняя продолжительность жизни — примерно 1213 лет, по некоторым источникам больные со стертой формой заболевания или удачно проведенными операциями по устранению дефектов развития иногда доживали до пятого-шестого десятка лет [1][2].
Клинический случай
Опекуны больного Р., 15.10.2000 г.р., впервые обратились за помощью в республиканскую детскую больницу г. Луганска с жалобами на задержку физического и умственного развития у ребенка, снижение аппетита, периодические судорожные припадки, вздутие и увеличение в объеме живота.
Из анамнеза жизни больного известно, что мальчик родился от третьей беременности, протекавшей на фоне злоупотребления алкоголем, III срочных родов, с массой тела 2800 г., длиной — 52 см, окружностью головы — 33 см, окружностью груди — 32 см, оценкой по шкале Апгар — 8. Период новорожденности протекал на фоне затянувшейся физиологической желтухи. Ребенок находился на грудном вскармливании до 2-х месяцев, затем — на искусственном (коровье молоко). На первом году жизни часто болел простудными заболеваниями (ОРВИ), вследствие чего ребенок получал вакцинацию с отставанием от календаря прививок.
Мальчик проживал в социально-неблагополучной семье, где мать и отец злоупотребляли алкоголем и бродяжничали.
В 4-летнем возрасте хирургами диагностирована декомпенсированная врожденная патология кишечника (мегадолихоколон) на фоне дистрофии III степени и гипертрихоза. Из-за частых ОРВИ только через 6 месяцев произведена левосторонняя гемиколэктомия по Ребейну.
Объективный статус: рост — 125,3 см (-5,1 сигмальных отклонений), масса тела — 11 кг 900 г. (ИМТ=7,4, 3 , левая — 2,05 см 3 .
Проведены консультации специалистов.
Окулист: ОИ — спокойны, глазное дно — диски зрительных нервов деколорированы, четкие, сосуды обычные. (Примечание: деколорирование дисков зрительных нервов встречается при различных наследственных заболеваниях и отражает начинающуюся атрофию ДЗН).
Генетик: кариотипирование — набор хромосом 46ХУ, половой хроматин — 0 %. Заключение: синдром Корнелии де Ланге, задержка психического развития.
Эндокринолог: задержка физического развития, нанизм, вторичный гипогонадизм.
Логопед: общее недоразвитие речи II-III уровня.
Сурдолог: двусторонняя нейросенсорная тугоухость II-III ст.
Нефролог: неполное удвоение левой почки.
Кардиолог: вторичная миокардиодистрофия, СН I.
Невролог: детский церебральный паралич резидуального генеза, стойкая ремиссия, стойкие выраженные двигательные расстройства, грубая задержка моторного и психоречевого развития. Врожденная гидроцефалия. Эпилептический синдром резидуального генеза.
Хирург: Врожденная аномалия толстого кишечника, гипокинетический тип, субкомпенсированная форма (СПО 2005 год). Вторичный хронический колит.
По результатам обследования установлен клинический диагноз: Синдром Корнелии де Ланге классическая форма. Задержка физического развития, нанизм генетически обусловленный. Задержка полового развития, вторичный гипогонадизм. Врожденная аномалия толстого кишечника, гипокинетический тип, субкомпенсированная форма (СПО 2005 г.). Вторичный хронический колит. Детский церебральный паралич, резидуального генеза, стойкая ремиссия, стойкие выраженные двигательные расстройства, грубая задержка моторного и психоречевого развития. Врожденная гидроцефалия. Эпилептический синдром резидуального генеза. Вторичная миокардиодистрофия, СН I. Неполное удвоение левой почки.
Главная задача терапии — увеличение продолжительности и улучшение качества жизни, дееспособности пациента, снижение проявлений симптоматики. Пациенту проводилась коррекция противосудорожными препаратами (депакин, финлепсин) миорелаксантами (мидокалм), ноотропными препаратами (фенибут), метаболическими средствами (рибоксин, аспаркам), ферментными препаратами (панкреатин), пробиотиками, гепатопротекторами, поливитаминами, анаболическими стероидами. Также назначался комплекс лечебной физкультуры, массаж.
В катамнезе в течение 2 лет наблюдения: ремиссия фебрильных судорог — 30 месяцев, частые острые респираторные инфекции до 6 раз в год, снижение слуха. Прогрессирование когнитивной недостаточности не отмечается.
Заключение
Приведенный клинический случай подтверждает необходимость проведения комплексного обследования детей с задержкой физического, псхоречевого и полового развития. Орфанные заболевания являются мультидисциплинарной проблемой, так как в патологический процесс вовлечены несколько органов и систем. Своевременная диагностика, лечение и коррекционные мероприятия позволят уменьшить клинические проявления, улучшить качество жизни и социальную адаптацию пациентов.
Список литературы
1. Козлова С.И. Демикова Н.С. Наследственные синдромы и медико-генетическое консультирование. – М.: КМК, Авторская академия; 2007.
3. Гинтер Е.К. Медицинская генетика. – М.: Медицина; 2003.
4. Жимулев И.Ф. Общая и молекулярная генетика. – Новосибирск: Сиб. унив. изд-во; 2002.
5. Бородулин В.И., Тополянский А.В. Синдромы и симптомы в клинической практике: эпонимический словарьсправочник. – М.: Эксмо; 2009.
6. Berney T.P., Ireland M., Burn J. Behavioral phenotype of Cornelia de Lange syndrome. // Arch. Dis. Child. – 1999. – Vol. 81. – P. 333-336.
Об авторах
Бугаенко Оксана Александровна – к.м.н, ассистент, кафедра педиатрии факультета последипломного образования.
Сиротченко Тамара Анатольевна – д.м.н, профессор, кафедра педиатрии факультета последипломного образования.
Бондаренко Галина Григорьевна – к.м.н, доцент, кафедра педиатрии факультета последипломного образования.
Вельковская Мария Марковна – врач-инфекционист.
Рецензия
Для цитирования:
For citation:
Контент доступен под лицензией Creative Commons Attribution 4.0 License.
Синдром Корнелии де Ланге (синдром Брахмана де Ланге, синдром дегенеративного нанизма амстердамского типа) представляет собой редкое заболевание, впервые описанное Брахманом в 1916 г., но носящее имя датского педиатра де Ланге. Синдром считается очень редким, частота его составляет всего один случай на 100000 новорожденных (Beck, 1976).
а) Патогенез. Синдром Корнелии де Ланге сопровождается мутациями гена NIBPL короткого плеча пятой хромосомы (Gillis et al., 2004; Krantz et al., 2004; Tonkin et al., 2004). Недавно было выявлено, что описанные мутации является основным этиологическим фактором данного синдрома и выявляются у 27-56% пациентов (Yan et al., 2006).
Тем не менее, среди пациентов с синдромом Корнелии де Ланге выявляется ряд других хромосомных аномалий (Jackson et al., 1993). Проявления, позволяющие выявить наличие данного синдрома, отмечаются при частичной трисомии дистальной части 3-й хромосомы (3q21-3ter) и других перестройках 3-й хромосомы (DeScipio et al., 2005). Синдром дупликации 3q хромосомы (dup (3q)-синдром) только на первый взгляд имитирует синдром де Ланге (Holder et al., 1994).
Данные изменения и изменения, связывающие ген МЕСР2 с синдромом Ретта, доказывают, что следует соблюдать осторожность, рассматривая ген NIBPL как единственную причину развития синдрома де Ланге.
Синдром де Ланге может наследоваться доминантным путем; в действительности все случаи синдрома представляют собой вновь возникшие мутации. Риск повторного рождения ребенка с данной патологией составляет 2-5%.
Синдром Корнелии де Ланге:
вдавленная переносица, низкая линия роста волос, сросшиеся брови,
короткие руки и согнутые пальцы рук и ног.
б) Клинические проявления. Синдром де Ланге является мультисистемным заболеванием, проявляющимся пре- и постнатальной задержкой роста, замедленным развитием, характерным дисморфизмом лица, мальформациями конечностей и множественными поражениями органов.
Кроме того, данное заболевание проявляется достаточно характерным поведенческим фенотипом, и в большинстве случаев серьезной или полной необучаемостью (Horsier и Oliver, 2006), часто сочетающейся с самодеструктивным поведением и избеганием социальных контактов, нередко достигая степени синдрома аутизма (Gillberg и Coleman, 2000; Arron et al., 2006; Bhuiyan et al., 2006).
Среди пациентов с мутациями отмечается тенденция к более выраженным изменениям массы тела, роста и средней окружности головы при рождении, дисморфизму лица и нарушению речи, чем среди пациентов, у которых мутации отсутствуют.
Читайте также: