
Синдром де груши реферат
Обновлено: 02.07.2024
В работе представлены варианты врожденного первичного гипогонадизма у мальчиков, показаны основные виды генетических нарушений полового развития, отмечены основные критерии дифференциальной диагностики различных вариантов врожденного первичного гипогонадизма, основанные на данных кариотипа, гормональных показателей, фенотипических особенностей при наиболее часто встречающихся нозологических формах данной патологии.
Clinical variants of congenital primary hypogonadism at boys
The paper presents variants of congenital primary hypogonadism at boys, shows the main types of genetic disorders of sexual development, marked the basic criteria of differential diagnosis of different variants of congenital primary hypogonadism based on data the karyotype, hormonal parameters, phenotypic characteristics with the most frequent nosological forms of this disease.
К гипогонадизму относят комплекс симптомов, отражающих недостаточность стероидогенной функции половых желез. Различают первичный (гипергонадотропный) и вторичный (гипогонадотропный) гипогонадизм. Первичные нарушения протекают с компенсаторной гиперсекрецией гонадотропных гормонов, вторичные — в условиях дефицита гонадотропной стимуляции. Такое разделение, основанное на содержании тестостерона и гонадотропных гормонов в крови, весьма условно, ибо в обоих случаях клинические проявления гипогонадизма не различаются. Вместе с тем картина гормональной недостаточности яичек, вплоть до формирования женского фенотипа у мужчины, может быть при нормальном биосинтезе андрогенов, если их рецепция нарушена. Дефицит гонадотропных гормонов может быть не только при заболеваниях гипофизарного уровня, но быть следствием избыточной продукции половых стероидов вне гонад либо экзогенного происхождения. В то же время разделение на первичный и вторичный гипогонадизм удобно в плане топической диагностики патологии.
Данные о распространенности синдрома гипогонадизма основаны на частоте выявления основных причин его развития. Так, анорхию наблюдают у 3-5% мальчиков с отсутствием яичек в мошонке. Синдром Клайнфельтера диагностируют у 1 из 500 мальчиков, а синдром Каллмана — у 1 из 5000. Гипогонадизм, связанный с другими причинами, встречается гораздо реже.
Классификация
В зависимости от уровня поражения гипоталамо-гипофизарной системы, различают следующие формы заболевания:
— гипергонадотропный (первичный) гипогонадизм;
— гипогонадотропный (вторичный) гипогонадизм;
— гипогонадизм, обусловленный резистентностью органов-мишеней.
В зависимости от времени возникновения:
В зависимости от этиологии:
• Первичный врожденный гипогонадизм
I. Нарушение развития гонад
Дисгенез семенных канальцев — синдром Клайнфельтера и его варианты
Синдром Клайнфельтера — распространенная патология (частота в мужской популяции 1 на 660 новорожденных мальчиков), является частой причиной гипогонадизма и бесплодия. Заболевание обусловлено наличием дополнительной Х хромосомы в кариотипе. В 90% случаев в кариотипе пациентов с синдромом Клайнфельтера выявляется одна дополнительная Х хромосома — 47, XXY. В остальных 10% имеются мозаицизм 46,XY/47,XXY, более выраженные X-хромосомные анэуплоидии (48,XXXY; 49,XXXXY, 47,XXY/48,XXXY) или структурно аномальные X-хромосомы.
Ключевым звеном в патогенезе синдрома Клайнфельтера является недостаточная продукция тестостерона клетками Лейдига (первичный или гипергонадотропный гипогонадизм). Основные клинические проявления данной патологии (и соответственно основные жалобы пациентов) — бесплодие, эректильная дисфункция, гинекомастия и избыточная масса тела. Фенотип таких пациентов разнообразный, как правило, они высокого роста, с евнухоидными пропорциями тела, маленькими плотными яичками. Отмечают также нарушения речи и снижение интеллекта.
Несмотря на высокую распространенность, эта патология более чем у половины больных на протяжении всей жизни остается нераспознанной. Более того, лишь у 10% пациентов диагноз устанавливают до периода полового созревания. Причины столь поздней диагностики могут быть обусловлены вариабельностью фенотипа, поздним клиническим проявлением симптомов заболевания, а также недостаточной осведомленностью врачей. Последствиями поздней постановки диагноза является несвоевременное назначение заместительной терапии препаратами тестостерона и как следствие того прогрессирование симптомов гипогонадизма и развитие осложнений, ассоциированных с гипогонадизмом. Наличие длительного персистирующего гипогонадизма у таких пациентов является моделью для изучения влияния дефицита андрогенов на половую функцию, предстательную железу, костную ткань, углеводный и жировой обмен, когнитивную функцию.
Аплазия герминативных клеток (синдром наличия только клеток Сертоли), или синдром Дель Кастильо
При гистологическом исследовании: малые размеры семенных канальцев, полное отсутствие сперматогенного эпителия при наличии сустентоцитов, нормальная базальная мембрана, клетки Лейдига.
При описываемом синдроме в отличие от синдрома Клайнфельтера отсутствует гинекомастия, отмечается нормальный мужской генотип 46 XY, проба на половой хроматин отрицательная. Копулятивные функции, спонтанные и адекватные эрекции, эякуляции, оргазм, половое влечение сохранены, больные не испытывают затруднений в половой жизни, жалуются только на бесплодие. В лечении гормонами не нуждаются. Прогноз в отношении способности к оплодотворению неблагоприятный. Лечение таким больным назначают только при наличии признаков недостаточной андрогенизации.
Анорхизм (Синдром исчезнувших яичек)
Этот синдром характеризуется отсутствием яичек. Мальчики рождаются с мужским фенотипом, со сформированными мужскими половыми органами, но яички в мошонке отсутствуют, что может расцениваться как крипторхизм. Кариотип 46 XY. Рост и развитие до полового созревания нормальные. В период полового созревания не развиваются вторичные половые признаки. Не происходит рост волос на лобке, в подмышечной области, на лице. Формируется евнухоидный тип телосложения. Может развиться остеопороз, иногда очень тяжелый. Половой член маленький, мошонка не развита, яичек в мошонке нет. При оперативном вмешательстве по поводу крипторхизма яичек не находят.
Полагают, что причиной исчезновения яичек является мутация гена тестис-стимулирующего фактора в коротком плече Y-хромосомы. Прекращение выработки этого фактора ведет к регрессии и исчезновению яичек. Предполагается, что регрессия и исчезновение яичек происходит после того, как у плода сформируется мужской фенотип, то есть спустя 70 и более дней после зачатия.
Крипторхизм
Крипторхизм означает отсутствие двух или одного яичка в мошонке. Он встречается у 5% мальчиков при рождении. У большинства из них яички опускаются в мошонку в течение первого года жизни. У 1% мальчиков крипторхизм сохраняется и в дальнейшем. В норме яички опускаются в мошонку между 20-й недели эмбрионального развития и родами.
Считается, что причиной крипторхизма может быть дефект в синтезе тестостерона и дегидротестостерона. Это подтверждено экспериментами, в которых было продемонстрировано, что дегидротестостерон необходим для нормального опускания яичек в мошонку. Яичко или яички могут находиться в паховом канале, и их нередко удается пальпировать. В других случаях оба или одно яичко находятся в брюшной полости.
В яичках уменьшено количество семенных канальцев и сперматогониев. Позже может развиться склероз ткани яичка. Если яички не опустились в мошонку до 4-5 лет. Возникает риск бесплодия. В 12% случаев у лиц с крипторхизмом, если яички находятся в брюшной полости, развиваются злокачественные опухоли неопустившегося яичка, чаще всего семиномы. Считают, что этому способствует необычная для яичек среда, более высокая температура в брюшной полости, чем в мошонке, которая является фактором риска развития новообразований яичка.
Синдром Ульриха-Нунан, или синдром Тернера у мужчин
Синдром рудиментарных яичек
Синдром рудиментарных яичек возникает в связи с тем, что внутренние половые органы образуются сразу из двух зачатков — мюллеровых и вольфовых протоков, причем первые образуют резко недоразвитую матку и маточные трубы, а вторые — рудиментарные яички и их придатки. Больные имеют евнухоидный вид, наружные половые органы — мужские, гипотрофированы; вторичные половые признаки недоразвиты. Рудиментарные яички нередко служат почвой для злокачественных опухолей.
Дисгенез гонад, или синдром Свайера
Аплазия клеток Лейдига
Причины аплазии клеток Лейдига не установлены. При этом дефекте яички сохранены, но в них отсутствуют клетки Лейдига, в которых синтезируется тестостерон. В отсутствии тестостерона не происходит должного развития семенных канальцев, и клетки Сертоли недостаточно синтезируют мюллеров — канал ингибирующий фактор. В связи с этим нарушается мужская дифференцировка и мальчик рождается с женским фенотипом — мужской псевдогермафродитизм. При рождении мальчика принимают за девочку. Однако кариотип мужской 46 XY.
Если гибель клеток Лейдига произошла после 10-12 недель эмбрионального развития, когда мужские органы уже сформировались, ребенок рождается с мужским фенотипом. В этих случаях заболевание диагностируется только в период полового созревания, когда не происходит развития вторичных половых признаков — отсутствует рост волос на лобке, в аксиллярных областях, на лице. Яички и половой член маленькие, мошонка не развита. Отсутствуют либидо и потенция. У лиц с мужским псевдогермафродитизмом, принятых за девочку, в период полового созревания имеет место первичная аменорея, не развиваются молочные железы, не происходит феминизация фигуры. При обследовании обнаруживается отсутствие матки и яичников. В паховом канале или в брюшной полости обнаруживают неразвитые яички. Кариотип XY.
Синдром 46ХХ у мужчин, или синдром де ля Шапеля
XX — нарушение или синдром половой реверсии — вариант синдрома Клайнфельтера. Клинические симптомы похожи, кроме более низкого роста и гипоспадии; случаи умственной неполноценности встречаются реже. Пациенты имеют кариотип 46ХХ. Такой парадокс объясняется экспрессией на клетках Н-Y антигена и предположительно наличием в составе генома структур Y-хромосомы.
Синдром ХYY
Фенотипические проявления XYY синдрома сходны с синдромом Клайнфельтера, но более вариабельны. Показатели спермы у этих мужчин варьируют от нормальных до азооспермии. Больные с наличием XXY-cиндрома отличаются высоким ростом, нередко мышечной силой, наличием угревой сыпи, а также снижением умственного развития и наклонностью к совершению криминальных действий. Симптомы гипогонадизма выражены умеренно (гипоплазированные тестикулы при нормальном половом члене). Оволосение на лице скудное. В спермограмме — олигозооспермия или у незначительной части больных — нормальный сперматогенез. У многих больных отмечается гипотония и замедленная речь. Содержание гонадотропинов умеренно повышено, а тестостерона в плазме — снижено. Метода лечения бесплодия не существует.
А.О. Поздняк
Казанская государственная медицинская академия
Поздняк Александр Олегович — доктор медицинских наук, профессор кафедры эндокринологии
Проведен анализ результатов полноэкзомного секвенирования больного задержкой психического развития и транзиторными психотическими состояниями. Выявлены участки делеции короткого плеча 18 хромосомы, включающие 57 генов и 12 некодирующих участков. Гены, наиболее значимые для развития заболевания у исследуемого пациента, были систематизированы по группам: регулирующие развитие эмбриона (TWSG1) и формирование плаценты (TGIF-1), развитие нервной системы и отдельных мозговых структур (CENT1, TGIF-1, ADCYAP1, ZBTB14, TWSG1), контроль транскрипции и внутриклеточный метаболизм, а также тканевой обмен. Показано, что генетический дефект в основном затрагивает этапы эмбрионального развития, нейрогенез и регуляцию транскрипции. Высказано предположение, что фенотически сходные психопатологические проявления имеют многовариантную генетическую предиспозицию в структуре коморбидности или синтропии.

1. Мутовин Г.Р. Нейроонтогенез и его нарушения / Г. Р. Мутовин, С. С. Жилина, З. Р. Умаханова // Детская больница — 2009. – № 2. — С. 36–43.
2. Пузырев В.П. Генетические основы коморбидности у человека / В. П. Пузырев // Генетика — 2015. — Т. 51. – № 4.– С. 491.
3. Гуткевич Е.В. Биологические парадигмы психических расстройств. Томские исследования: история, реальность и перспективы / Е. В. Гуткевич, Е. А. Гуткевич // Сибирский вестник психиатрии и наркологии — 2016. — № 4(93). — С. 14–15.
4. Blair D.R., Lyttle C.S., Mortensen J.M., Bearden C.F., Jensen A.B. et al. A nondegenerate code of deleterious variants in mendelian loci contributes to complex disease risk // Cell. 2013. Vol. 155. № 1.P. 70–80.
5. Andler W., Heuveldop A., Polichronidou T. Endokrinologische storungen bei Deletionen des chromosomes 18 // Monatsschr. Kinderheilkd. 1992. № 140.5. P. 303–306.
7. Babovic-Vuksanovic D., Jenkins S.C., Ensenauer R., Newman D.C., Jalal S.M. Subtelomeric deletion of 18p in an adult with paranoid schizophrenia and mental retardation // Am. J. Med. Genet. A 2004. Vol. 124. № 3. P. 318–322.
8. Wester U., Bondeson M.L., Edeby C., Annerén G. Clinical and molecular characterization of individuals with 18p deletion: A genotype-phenotype correlation // Am. J. Med. Genet. Part A. 2006. Vol. 140. № 11. P. 1164–1171.
9. Wang X. et al. Asymmetric centrosome inheritance maintains neural progenitors in the neocortex // Nature. 2009. Vol. 461. № 7266. P. 947–955.
10. Inooka H. et al. Conformation of a peptide ligand bound to its G-protein coupled receptor // Nat. Struct. Biol. 2001. Vol. 8. № 2. P. 161–165.
11. Bertolino E., Reimund B., Wildt-Perinic D., Clerc R.G. A novel homeobox protein which recognizes a TGT core and functionally interferes with a retinoid-responsive motif // J. Biol. Chem. 1995. Vol. 270. № 52. P. 31178–31188.
12. Gripp K.W. et al. Mutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determination. // Nat. Genet. 2000. Vol. 25. № 2. P. 205–208.
13. Graf D. et al. Evolutionary conservation, developmental expression, and genomic mapping of mammalian Twisted gastrulation // Mamm. Genome. 2001. Vol. 12. № 7. P. 554–560.
14. Brehm J.M. et al. Stress and bronchodilator response in children with asthma // Am. J. Respir. Crit. Care Med. 2015. Vol. 192. № 1. P. 47–56.
15. Tamas A. et al. Effect of PACAP in Central and Peripheral Nerve Injuries // Int J Mol Sci. 2012. Vol. 13. № 7. P. 8430–8448.
16. Stevens J.S. et al. PACAP receptor gene polymorphism impacts fear responses in the amygdala and hippocampus // Proc. Natl. Acad. Sci. U. S. A. 2014. Vol. 111. № 8. P. 3158–3163.
17. Wang L. et al. PAC1 receptor (ADCYAP1R1) genotype is associated with PTSD’s emotional numbing symptoms in Chinese earthquake survivors // J. Affect. Disord. 2013. Vol. 150. № 1. P. 156–159.
18. Hashimoto R. et al. Possible association between the pituitary adenylate cyclase-activating polypeptide (PACAP) gene and major depressive disorder // Neurosci Lett. 2010. Vol. 468. № 3. P. 300–302.
19. Katayama T., Hattori T., Yamada K., Matsuzaki S., Tohyama M. Role of the PACAP-PAC1-DISC1 and PACAP-PAC1-stathmin1 systems in schizophrenia and bipolar disorder: novel treatment mechanisms? // Pharmacogenomics. 2009. Vol. 10. № 12. P. 1967–1978.
20. Satoh K. et al. DAP-1, a novel protein that interacts with the guanylate kinase-like domains of hDLG and PSD-95 // Genes Cells. 1997. Vol. 2. № 6. P. 415–424.
21. Pickard B.S. et. al. Candidate psychiatric illness genes identified in patients with pericentric inversions of chromosome 18 // Psychiatr. Genet. 2005. Vol. 15. № 1. P. 37–44.
22. Stewart S.E. et al. Genome-wide association study of obsessive-compulsive disorder // Mol. Psychiatry. 2013. Vol. 18. № 7. P. 788–798.
23. Benit P., Beugnot R., Chretien D., Giurgea I. et al. Mutant NDUFV2 subunit of mitochondrial complex I causes early onset hypertrophic cardiomyopathy and encephalopathy // Hum. Mutat. 2003. Vol. 21. № 6. P. 582–586.
24. Loeffen J.L et al. cDNA of eight nuclear encoded subunits of NADH:ubiquinone oxidoreductase: human complex I cDNA characterization completed. // Biochem. Biophys. Res. Commun. 1998. Vol. 253. № 2. P. 415–422.
25. Sobek-Klocke I. et al. The human gene ZFP161 on 18p11.21-pter encodes a putative c-myc repressor and is homologous to murine Zfp161 (Chr 17) and Zfp161-rs1 (X Chr) // Genomics. 1997. Vol. 43. № 43. P. 156–164.
26. Washizuka S. et al. Association of mitochondrial complex I subunit gene NDUFV2 at 18p11 with bipolar disorder in Japanese and the National Institute of Mental Health pedigrees // Biol. Psychiatry. 2004. Vol. 56. № 7. P. 483–489.
27. Washizuka S. et al. Association of mitochondrial complex I subunit gene NDUFV2 at 18p11 with bipolar disorder // Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2003. Vol. 120B. № 1. P. 72–78.
28. Robinson B.H. Human Complex I deficiency: Clinical spectrum and involvement of oxygen free radicals in the pathogenicity of the defect // Biochim. Biophys. Acta - Bioenerg. 1998. Vol. 1364. № 2. P. 271–286.
29. Lee K. Human zinc finger protein 161, a novel transcriptional activator of the dopamine transporter // Biochem. Biophys. Res. Commun. 2004. Vol. 313– P. 969–976.
30. Orlov S.V. et al. Novel repressor of the human FMR1 gene - Identification of p56 human (GCC)n-binding protein as a Kruppel-like transcription factor ZF5 // FEBS J. 2007. Vol. 274. № 18. P. 4848–4862.
31. Almen M., Nordstrom K.J., Fredriksson R., Schioth H.B. Mapping the human membrane proteome: a majority of the human membrane proteins can be classified according to function and evolutionary origin // BMC Biol. 2009. Vol. 7 № 1. P. 50.
32. Senyürek I. et al. Processing of laminin α chains generates peptides involved in wound healing and host defense // J. Innate Immun. 2014. Vol. 6. № 4. P. 467–484.
33. Aldinger K.A., Mosca S.J., Tetreault M., Dempsey J.C., Ishak G.E., Hartley T. et al. Mutations in LAMA1 cause cerebellar dysplasia and cysts with and without retinal dystrophy // Am. J. Hum. Genet. 2014. Vol. 95. № 2. P. 227–234.
34. Wetherill L. et al. Association of substance dependence phenotypes in the COGA sample // Addict. Biol. 2015. Vol. 20. № 3. P. 617–627.
36. Volkers L., Mechioukhi Y., Coste B. Piezo channels: from structure to function // Pflugers Arch. Eur. J. Physiol. 2015. Vol. 467. № 1. P. 95–99.
37. McMillin M.J. et al. Mutations in PIEZO2 cause Gordon syndrome, Marden-Walker syndrome, and distal arthrogryposis type 5 // Am. J. Hum. Genet. 2014. Vol. 94. № 5. P. 734–744.
Мы провели комплексный, клинико-патогенетический анализ результатов полноэкзомного секвенирования пациента с гетерозиготной делецией 18 хромосомы. Особенностью клинических проявлений данного испытуемого было наличие у него нетипичных для данного синдрома транзиторных психопатологических состояний шизоформного характера.
Цель исследования заключалась в изучении молекулярно-генетических предпосылок развития заболевания.
Задачи исследования включали:
1. Определение генов, попавших в зону повреждения, и их роли в регуляции функционирования организма.
2. Систематизация указанных генов с учетом их значимости для нейроонтогенеза и риска развития заболеваний ЦНС.
3. Исследование характера и генетической структуры коморбидности выявленных психопатологических проявлений.
Материалы и методы исследования
Анализ проведен методами секвенирования следующего поколения с использованием наборов для обогащения экзома Genotek Clinical Exome (Illumina Inc., США) и секвенирования ДНК производства Illumina (Illumina Inc., США). Прибор - Illumina HiSeq2500. Среднее покрытие - 78. Биоинформатическая обработка данных произведена в соответствии с регуляциями ACMG (США). Референсная последовательность Human genome 19 (hg19) build 37.
Выписка из амбулаторной истории болезни испытуемого.
Из анамнеза: родился первым ребенком из 3. Возраст матери при его рождении – 32 года, отцу – 35 лет. Роды в срок, вес при рождении 3,250 г. Младшие дети здоровы. С детства отставал в психическом развитии от сверстников. Фразовая речь с 5 лет. Ходить начал с 1 года. В связи с отставанием в умственном развитии посещал коррекционные занятия. С 8 лет был переведен на индивидуальное обучение. Умеет читать и писать. Всегда отличался покладистым характером, не конфликтовал с другими детьми, хотя они его и обижали. Физически развит хорошо, на момент обследования рост 163 см, но медлителен, моторно неловок. С удовольствием катается на велосипеде. В возрасте 12 лет был обследован в Медико-генетическом научном центре РАМН. Кариотип: 46,XY, del (18)(p11.2). Осенью 2014 года резко изменился в поведении: стал сопротивляться выполнению привычных занятий, отказывался готовить уроки, не реагировал на обращенную к нему речь, что-то бормотал, стереотипно повторял одни и те же фразы без связи с окружающей обстановкой. Плохо спал. Если раньше писал разборчиво и четко, то теперь – какие-то каракули. Через 1,5 месяца указанные нарушения поведения купированы (в течение 2 недель принимал пантокальцин и мексидол). Очередное обострение заболевания началось в конце мая 2015 года. Снова замкнулся, сначала перестал выходить на улицу, потом и из своей комнаты. Ухудшение психического состояния постепенно нарастало: перестал реагировать на обращенную речь, не спал по ночам, отказывался от контакта с родителями, стереотипно повторял бессмысленные фразы. Отмечалось повышение аппетита.
Осмотрен 07 июля 2015 г. Со слов родителей больного: ночью плохо спит, бродит по комнате. Не реагирует на окружающих, не может самостоятельно одеться, принять пищу – только с помощью близких. Отсутствует чувство насыщения.
Заключение ЭЭГ-исследования (11.02.2016). Отмечаются отчетливые диффузные изменения биоэлектрической активности мозга регуляторного характера с признаками раздражения коры, подкорково-диэнцефальных и медиобазальных структур в виде десинхронизации, снижения индекса альфа-активности, значительного увеличения индекса диффузной и синхронной медленно-волновой активности в фоновой записи. Снижение межполушарной интеграции в быстром частотном диапазоне преимущественно в передних отделах (по сравнению с показателями банка данных возрастной нормы).
В результате секвенирования клинического экзома у пациента были выявлены структурные нарушения генома – гетерозиготная делеция короткого плеча 18 хромосомы, локусы p11.32, p11.31, р11.23 и р11.22 (рис. 1). Зона делеции включала суммарно 268 экзонов (-11269401п.н.). Начальная позиция делеции в геноме – 10 500 п.н., конечная позиция – 11287773 п.н.


Рис. 1. Зона делеции 18 хромосомы испытуемого
В зоне делеции оказалось 57 генов и 14 некодирующих участков. Основываясь на литературных данных, был проведен анализ роли и значения каждого из указанных генов для развития и жизнедеятельности организма. В соответствии с задачами исследования, в отдельную группу выделены гены, имеющие наиболее существенное значение для развития заболевания (таблица).
Клиника. Низкая масса тела при рождении, профиль плоский или вогнутый. Маленький рост, микроцефалитная форма черепа, круглое лицо, высокое небо (иногда с расщелиной). Очаги облысения на голове либо тотальная алопеция. Характерна деформация зубов и ушных раковин, пупочечные и паховые грыжи. Аномалия кистей рук и пальцев, синдактилия пальцев ног. У мальчиков часто недоразвитие половых органов. Отмечается умственная отсталость.
Патогенез. У больных с грубой мозговой патологией резкое снижение продолжительности жизни. Косоглазие, мышечная гипотония, гипоплазия полового члена и мошонки у мальчиков и гипоплазия малых половых губ у девочек. Пороки сердца, иногда почек.
Диагностика. Исследование кариотипа, цитологическое обследование.
Частота. 1: 60000.
Синдром Лежена
Патогенез. Пороки сердца, иногда почек. Пороки развития зрительной системы.
Диагностика. Дерматоглифика (повышенное число завитков на пальцах рук), цитологическое обследование.
Синдром Реторе
Причина. Частичная трисомия по короткому плечу хромосомы 9.
Патогенез. Глазные аномалии, у 25%- врожденные пороки сердца. Двигательные расстройства, нарушение координации.
Диагностика. Дерматоглифика, цитологическое обследование, кариологическое исследование.
Синдром Брадера – Вилли
Причина. Утраивается участок 15-й хромосомы отцовского происхождения
Клиника. Мышечная гипотония, половое недоразвитие, ожирение, умственная отсталость. Низкая масса тела при рождении, пониженная температура. Потом развивается чрезвычайный аппетит. Деформированные низко расположенные ушные раковина и мягкие ушные хрящи, подковообразная форма рта, короткая губа, неправильный рост зубов. Диспропорциональность стоп и кистей. Нарушение осанки.
Патогенез. Частые грыжи, патология кистей и стоп. В пубертатном возрасте диабет. Взрослые страдают гиперсомнией, ишемической болезнью сердца, инфаркт миокарда.
А.Г. Шаповалов 1 , врач-кардиоревматолог, С.Е. Онищенко 2 , врач-генетик, О.Б. Полодиенко 1 , канд. биол. наук, генетик. 1 Городской детский лечебно-диагностический центр им. ак. Б.Я. Резника, г. Одесса. 2 Областная детская клиническая больница, г. Херсон
Содержание статьи:
Генетически обусловленная патология составляет значительную долю в структуре перинатальной смертности, детской заболеваемости и инвалидности. Известно, что частота хромосомных аномалий среди новорожденных составляет 0,7-0,8%, у мертворожденных – 5%, в материале спонтанных выкидышей – 50%. Ведущая роль в выявлении хромосомной этиологии заболевания принадлежит цитогенетическим методам исследования.
Раннее цитогенетическое, а при необходимости – молекулярно-цитогенетическое, обследование детей с врожденными пороками развития (ВПР) важно для установления диагноза, определения семейных форм перестроек, позволяет врачу выбрать своевременные методы медицинской коррекции и социальной адаптации ребенка.
Согласно литературным данным, моносомия короткого плеча хромосомы 18 (del(18)(p11)) (частичная моносомия по короткому плечу хромосомы 18, парциальная моносомия короткого плеча хромосомы 18, 18р-синдром, синдром де Груши, код по ОМIM – 14639) выделена в отдельный клинический синдром с четко определенными признаками [17] и критическим районом 18р11.1-11.21 [33]. Частота встречаемости заболевания составляет 1 на 50000 новорожденных с соотношением женщин и мужчин 3:2 [3, 26, 33]. Первыми описали пациента с делецией по короткому плечу хромосомы 18 де Груши с сотрудниками в 1963 г. [7], а к 2008 г. в мировой литературе присутствовала информация приблизительно о 150 случаях болезни [11, 24].
Цель работы – уточнение диагноза заболевания у ребенка с отставанием в физическом, психомоторном и умственном развитии.
Материалы и методы исследования
Проведено комплексное клинико-генеалогическое и цитогенетическое обследование пробанда С. с использованием параклинических и инструментальных методов. Клиническое и медико-генетическое консультирование ребенка и его семьи осуществлено врачом-генетиком областной детской клинической больницы (г. Херсон), кариотипирование проведено в лаборатории клинической молекулярно-генетической диагностики ГДБ им. ак. Б. Я. Резника (г. Одесса).
Для кариотипирования использовали метафазные хромосомы из лимфоцитов периферической крови пробанда, полученные согласно стандартной методике [10], с последующим окрашиванием хромосом GTG- и С-методами. Идентификацию хромосом осуществляли в соответствии с международной системой цитогенетической номенклатуры ISCN (An international system for human cytogenetic nomenclature, 2016) [2].
Результаты исследования и их обсуждение
Пробанд С. – девочка от вторых родов, второй беременности (первые роды завершились рождением здоровой девочки). На момент родов матери 31 год, отцу – 27 лет; родители соматически здоровы; не состоят в родстве; условия труда не связаны с вредными производственными факторами. По линии матери родословная отягощена выкидышами (привычное невынашивание беременности у бабушки), сахарным диабетом 1 типа, бронхиальной астмой.
Беременность протекала с гестозом первой половины, угрозой прерывания на 5-м месяце, кровомазанием и перенесением в 24-й неделе острой респираторной инфекции (ринит). Биохимический скрининг 1-2-го триместра беременных не проводился. Пренатальное УЗИ плода осуществлялось 4 раза, патология не выявлялась. Роды в сроке 39-40 недель гестации, в головном предлежании плода. Масса тела ребенка при рождении 3000 г, оценка по шкале Апгар – 7-8 баллов. Закричала сразу. Выписана из роддома на 3-и сутки.
Результаты клинических и генетических исследований пробанда
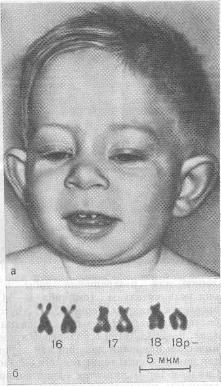
Участковым педиатром были обнаружены фенотипические маркеры вероятных аномалий эмбрионального развития, в связи с чем в возрасте 6 мес 3 нед девочка была проконсультирована врачом–клиническим генетиком. Выявлены множественные стигмы дизэмбриогенеза: монголоидный разрез глаз, эпикант, широкая запавшая переносица, диспластические низко расположенные ушные раковины, форма лица с уплощенной спинкой маленького носа, глубокопосаженными глазными яблоками, тонкими и опущенными вниз губами (рот карпа).
/images/DL19-1_3437_r1-300x228.jpg)
Рис. 1. Кариограмма пробанда С. GTG-метод окрашивания хромосом
/images/DL19-1_3437_r1.jpg)
Рис. 1. Кариограмма пробанда С. GTG-метод окрашивания хромосом
Для проведения комплексного обследования ребенок был госпитализирован в неврологическое отделение многопрофильного педиатрического стационара. Результаты диагностических исследований документировали наличие расходящегося содружественного косоглазия слева, ассиметричную вентрикулодилатацию по данным НСГ. Патологии при осмотре кардиолога (ЭКГ, ЭхоКГ), УЗИ органов брюшной полости, отклонений в гормональной панели скринига функции щитовидной железы, а также ортопедических и сурдологических заболеваний выявлено не было. Установлена умеренная задержка развития психических и речевых функций, наличие синдрома двигательных расстройств (мышечная дистония с тенденцией к гипотонии). В результате цитогенетического обследования во всех проанализированных метафазных пластинках (30) ребенка выявлена моносомия по короткому плечу хромосомы 18 – кариотип 46, XX, del(18)(p11.1)(рис.1).
Происхождение аберрантной хромосомы (спорадическое или унаследованное от одного из родителей носителя сбалансированной перестройки) установить не удалось, так как родители от кариотипирования отказались.
На основании полученных данных у ребенка была диагностирована хромосомная болезнь – делеция короткого плеча хромосомы 18. Пациент динамически наблюдается профильными специалистами, проводится комплекс лечебных, психокорректирующих и социально-реабилитационных мероприятий.
Варианты кариотипов
Известны случаи как тотальной, так и частичной делеции короткого плеча хромосомы 18 [17]. У большинства больных (85%) моносомия по короткому плечу хромосомы 18 – результат мутации de novo [23]. Описания семейных случаев немногочисленны [15, 21, 27, 28, 30, 32], причем отмечается вариабельность клинической картины заболевания у членов одной семьи с делецией короткого плеча хромосомы 18 [4]. Парциальная моносомия короткого плеча хромосомы 18 у ребенка может быть также следствием наличия в кариотипе одного из родителей либо клона клеток с 18р-, либо сбалансированной транслокации с участием короткого плеча этой хромосомы.
Так, в публикации [30] сообщается о семье, в которой у матери был мозаичный кариотип по делеции короткого плеча хромосомы 18, а у обоих ее детей во всех клетках идентифицировали моносомию короткого плеча этой хромосомы. Сочетанная хромосомная патология – моносомия по короткому плечу хромосомы 18 и трисомия по короткому плечу хромосомы 16 (46, ХY, der(18) t(16;18)(p11.2; p11.2) pat обнаружена у ребенка отца со сбалансированной транслокацей в кариотипе – 46, ХY, t(16;18)(p11.2; p11.2) [19]. Частичная делеция короткого плеча хромосомы 18 установлена у ребенка, мать которого имела сбалансированную транслокацию части короткого плеча хромосомы 18 на короткое плечо хромосомы 17, причем идентичную перестройку имели ее сестра и отец [13].
Особый интерес, с точки зрения влияния на онтогенез нарушения сбалансированности генома при одновременной утрате и приобретении различного генетического материала хромосомы 18, представляют публикации [1, 8, 22]. Сообщается о детях, в кариотипе которых обнаружено два клона клеток – один с частичной моносомией по короткому плечу хромосомы 18 (46, ХХ, del(18)(p11)), другой – с изохромосомой по длинному плечу хромосомы 18 (46, ХХ, i(18)(q10)). Особенности кариотипа обусловили у пробандов наличие совокупности фенотипических признаков, присущих синдромам моносомии 18р и трисомии 18q.
Клиническая картина
Нежизнеспособные дети с частичной моносомией по короткому плечу хромосомы 18, погибшие в первые дни или месяцы жизни, имели грубые врожденнные аномалии мозга и лицевого скелета (аринанцефалию, циклопению, расщелины губы и неба, уродства носового скелета); множественные пороки развития желез внутренней секреции (аплазию гипофиза; гипоплазию или дисплазию щитовидной железы и надпочечников); нарушения формирования селезенки и других внутренних органов [6, 9, 20, 30].
Несмотря на широкую вариабельность дисморфий при синдроме моносомии по короткому плечу хромосомы 18, есть ряд симптомов, встречающихся с устойчивым постоянством, к числу которых относится общее физическое недоразвитие [29]. Возраст пробанда не позволяет с уверенностью верифицировать наличие гипостатуры и короткой шеи, так как ситуация требует динамического наблюдения. Низкой линии роста волос у ребенка не документировано.
Весьма типичны маркеры окуло- и краниофациального дисморфизма, представленные в рассматриваемом случае страбизмом, эпикантом, диспластичными, увеличенными в размерах ушными раковинами, уплощенной переносицей, широким ртом с тонкими губами [25, 29]. В то же время не отмечено наличие птоза, экзофтальма, нистагма, гипертелоризма, нередко описываемых в литературных источниках в контексте синдрома де Груши [14, 17].
Часто встречающимся признаком являются разнообразные дентальные дефекты (аномалии количества зубов, кариес, сопутствующее готическое небо) [18, 29].
Описаны ассоциации заболевания с эндокринными расстройствами (гипотиреоидизм, диабет); повышенным риском развития аутоиммунной патологии в виде ревматоидного артрита; дефицитом IgA [17], кератодерматозами [5]; врожденной гипоглоссией [12]; врожденными пороками сердца (в 10% наблюдений), в том числе синдромом гипоплазии левого сердца [31].
Таким образом, клинические проявления рассматриваемого синдрома у пробанда в целом соответствуют картине, описанной в литературе, и требуют динамического наблюдения для уточнения степени последующей презентабельности, в том числе выраженности интеллектуального дефицита.
Выводы
- Комплексное медико-генетическое обследование пробанда позволило в раннем возрасте выявить редкое хромосомное заболевание, отличающееся полиморфизмом клинических проявлений.
- Своевременная диагностика обеспечила формирование индивидуализированного мультидисциплинарного подхода к медикаментозной терапии проявлений нозологии наряду с поведенческой коррекцией, психосоциальной реабилитацией и адаптацией.
- Актуализирована важность интегративного взаимодействия и преемственности медико-генетической, акушерско-гинекологической и педиатрической служб в свете возрастания связующей роли медицинской генетики в общей системе профилактики болезней человека.
Список литературы
1. Бадалян Л. О. Редкий случай мозаицизма по хромосоме 18, кариотип: 46, ХХ, del(18)(p11)/46, XX, i(18q) / Бадалян Л. О., Мутовин Г. Р., Малыгина Н. А., Петрухин А. С. Генетика. 1983. Т. 19, № 11. С. 1912-1915.
2. Зерова-Любимова Т. Е., Горовенко Н. Г. Цитогенетичні методи дослідження хромосом людини (методичні рекомендації). К., 2003. 23 с.
5. Carvalho C. A. Keratosis pilaris and ulerythema ophryogenes in a woman with monosomy of the short arm of chromosome 18 / Carvalho C. A., Carvalho A. V.E., Kiss A., Paskulin G., Götze F. M. An Bras Dermatol. 2011. V. 86 (4 Supl. 1). P. 42-45.
6. Fiant S. Lewis T. Presumptive deletions of the short arm of chromosome 18 in a cyclops. Human Chromos. Newsletter. 1964. V. 14(5).
8. Ghaziuddi M. Abnormalities of chromosome 18 in a girl with mental retardation and autistic disorder / Ghaziuddi M., Sheldon S., Tsai L. Y., Alessi N. A. Journal J. Intellectual Disability Research. 1993. V. 37. P. 313-317.
9. Haworth J. C., Melody H., Lewis A. J. Cebocephaly with endocrine dysgenesis (report of 3 case). Pediatrics. 1961. V. 37, № 59. P. 726.
10. ISCN. An international system for human cytogenomic nomenclature // Eds. J. McGowan-Jordan J., Simons A., Schmid M. Basel: Karqer, 2016. 139 р.
11. Kim Y. M. Del (18p) syndrome with increased nuchal translucency in prenatal diagnosis / Kim Y. M., Cho E. H., Kim J. M. et al. Prenat. Diagn. 2004. V. 24. Р. 161-164.
12. Klaphake S. A patient with chromosome 18p deletion and congenital hypoglossia / Klaphake S., van Doon M. F., Sender R. E.M et al. Clin Dysmorphol. 2018. Apr. V. 27(2). P. 46-48.
14. Li-Juan Xu. A case of 18p deletion syndrome / Li-Juan Xu, Ly-Xian Wu, Oing Yuan et al. Int Med Case Rep. S. 2017. V. 10. P. 15-18.
15. Maranda B., Lemieux N., Lemyre E. Familial deletion 18p syndrome: case report. BMC Medical. Genetics. 2006;7:60. doi:10.1186/1471-2350–7-60.
16. McDermott A. Archinencephaly associated with a deficiency involving chromosome 18 / McDermott A., Insley J., Barton M.E et al. J. Med. Genet. 1968. V. 5, № 1. P. 60.
17. Minire Hasi-Zogaj. A review of 18p deletions / Minire Hasi-Zogaj, Courtney Sebold, Heard P. et al. American J. of Med. Genet. in Part (Seminars in Medical Genetics). 2015. V. 169. P. 251-264.
18. Naudi A. B., Fung D. E. A child with 18p- syndrome: a case report. Spec. Care Dentist. 2007. Jan-Feb. V. 27(1). P. 12-14.
19. Nicole Schmidt. Developmental delay and multiple congenital anomalies in a child with a unique combination of partial monosomy 18 and partial trisomy 16 / Nicole Schmidt, Don C Van Dyke, Kim M. D. et al. Developmental Medicine & Child Neurology. 2001. V. 43. P. 130-132.
20. Nitowsky H. M. Partial monosomy in the cyclops malformation / Nitowsky H. M., Sindhvananda N., Konigsberg U. R. et al. Pediatrics. 1966. V. 59, № 2. P. 260.
21. Rigola M. A. Characterization of a heritable partial monosomy 18p by molecular and cytogenetic analysis / Rigola M. A., Plaja A., Mediano C. et al. Am J. Med. Genet. 2001. V. 104(1). P. 37-41.
22. Peng D. A study of a rare chromosomal disorder mosaic 46, XX, del(18)(p11.2)/46, XX, i(18q) / Peng D., Long P. P., Wen B., Yu R. H. J. Genet. 2013. V. 92. № 2. P. 611-615.
23. Spinner N. B., Emanuel B. S. Deletions and other structural abnormalities of the autosomes. In Emery and Rimoin’s principles and practice of medical genetics Volume 1. 4th/edition. Edited by: Rimoin D. L., Emery A. E.H. Londo; New York, Churchill Livingstone. 2002. Р. 1210-1211.
24. Schaub R. L. Molecular characterization of 18p deletions: evidence for a breakpoint cluster / Schaub R. L., Reveles X. T., Baillargeon J. et al. Genet. Med. 2002. V. 4. P. 15-19.
25. Sun H. Genotype-Phenotype Analysis, Neuropsychological Assessment, and Growth Hormone Response in a Patient with 18p Deletion Syndrome / Sun H., Wan N., Wang X. et al. Cytogenet Genome Res. 2018. V. 154(2). P. 71-78. doi: 10.1159/000487371. Epub 2018 Mar 16.
26. Thompson R. W., Peters J. E., Smith S. D. Intellectual, behavioural, and linguistic characteristics of three children with 18p-syndrome. J. Dev Behav. Pediatr. 1986. V. 7. Р. 1-7.
27. Tonk V., Krishna J. Case report: de novo inherited 18p deletion in a mother-fetus pair with extremely variable expression, confirmed by fluorescence in situ hybridization (FISH) analysis. Eur. J. Obstet. Gynecol. Reprod. Biol. 1997. V. 73(2). P. 193-196.
28. Tsukahara M. Familial Del(18p) syndrome / Tsukahara M., Imaizumi K., Fujita K. et al. Am J. Med. Genet. 2001. V. 99(1). P. 67-69.
29. Turleau C. Orphanet Journal of Rare Diseases. 2008;3:4. doi:10.1186/1750-1172–3-4.
30. Uchida I. A. Familial short arm deficiency of chromosome 18 concomitant with arhinencephaly and alopecia congenital / Uchida I. A., McRae H.C., Wang H. C., Ray M. Am J. Hum. Genet. 1965. V. 17(5). P. 410-419.
31. Vasquez J. C. Hypoplastic left heart syndrome with intact atrial septum associated with deletion of the short arm of chromosome 18 / Vasquez J. C., Rabah R., Delius R. E. et al. Cardiovascular Pathol. 2003 Mar-Apr; 12(2). P. 102-104.
32. Velagaleti G. V. Familial deletion of chromosome 18 (p11.2) / Velagaleti G. V., Harris S., Carpenter N. J. et al. Ann Genet. 1996. V. 39(4). P. 201-204.
33. Wester U. Clinical and molecular characterization of individuals with 18p deletion: a genotypephenotype correlation / Wester U., Bondeson M., Edeby C. et al. Am J. Med. Genet A. 2006. V. 140. Р. 1164-1171.
КЛІНІЧНИЙ ВИПАДОК МОНОСОМІЇ ПО КОРОТКОМУ ПЛЕЧУ ХРОМОСОМИ 18
А.Г. Шаповалов 1 , С.Е. Онищенко 2 , О.Б. Полодієнко 1
1 Міський дитячий лікувально-діагностичний центр ім. Б.Я. Резника, м. Одеса,
2 Обласна дитяча клінічна лікарня, м. Херсон
Резюме
В статті розкрита важливість раннього цитогенетичного обстеження дітей з вродженими вадами розвитку, наведений клінічний випадок моносомії по короткому плечу хромосоми 18, розглянуті варіанти каріотипу при цій патології та її можливі фенотипові прояви.
Ключові слова: хромосомні захворювання, вроджені хвороби, генетичні хвороби.
CLINICAL CASE OF SHORT ARM DEFICIENY OF CHROMOSOME18
A.G. Shapovalov 1 , S.E. Onischenko 2 , O.B. Polodienko 1
1 Reznik City Children’s Medical Center, Odessa,
2 Herson Regional Children’s Clinical Hospital
Abstract
The article reveals the importance of early cytogenetic examination of children with congenital anomalies. It presents a clinical case of 18p– syndrome and describes it’s karyotype and phenotypic manifestations.
Key words: chromosome disorders, congenital diseases, genetic diseases.
Раздел только для специалистов в сфере медицины, фармации и здравоохранения!

А.В. Дегтярева 1,2 , д.м.н., А.А. Пучкова 1 , к.м.н., А.В. Болмасова 1 , к.м.н., М.А. Меликян 3 , к.м.н.
1 Научный центр акушерства, гинекологии и перинатологии им. академика В.И. Кулакова Минздрава России, Москва
2 Первый Московский государственный медицинский университет им. И.М. Сеченова Минздрава России
3 Эндокринологический научный центр Минздрава России, Москва
Синдром моносомии 18р- – крайне редкое заболевание (1:50000 новорожденных, родившихся живыми). Врожденный гипопитуитаризм является одним из проявлений данного синдрома в 13% случаев. Редкость данной патологии обуславливает трудности ранней диагностики врожденного гипопитуитаризма в силу недостаточной осведомленности среди педиатров и неонатологов. В данной статье представлен клинический случай врожденного гипопитуитаризма у девочки с синдромом моносомии 18р-, который проявлялся после рождения симптомами холестаза и гипогликемией.
A.V. Degtyareva 1,2 , MD, А.А. Puchkova 1 , PhD in medicine, A.V. Bolmasova 1 , PhD in medicine, M.A. Melikyan 3
1 Kulakov Scientific Center for Obstetrics, Gynaecology and Perinatology of the Ministry of Health, Moscow
2 Sechenov First Moscow State Medical University of the Ministry of Health of Russia
3 Endocrinology Research Center of the Ministry of Health of Russia, Moscow
Cholestasis, hypoglycemia and unusual phenotype as the manifestations of congenital hypopituitarism as part of monosomy 18P- syndrome
The monosomy 18p-syndrome refers to an extremely rare disorder (1:50,000 live-born infants). Congenital hypopituitarism is one of the manifestations of this syndrome in 13% of cases. The rarity of this pathology causes difficulties in the early detection of congenital hypopituitarism due to lack of awareness among paediatricians and neonatologists. The article presents a clinical case of congenital hypopituitarism in a girl with monosomy 18p-syndrome, which manifested itself after birth in the form of cholestasis and hypoglycaemia.

Список литературы
1. de Grouchy J, Lamy M, Thieffry S, Arthuis M, Salmon CH: Dysmorphie complexe avec oligophrenie: deletion des bras courts d’un chromosome 17-18. C R Acad Sci, 1963, 258: 1028.
2. de Grouchy J. The 18p, 18q and 18 syndromes. Birth defects Orig Art Ser, 1969, V: 74-87.
3. Nusbaum C, Zody MC, Borowsky ML, Kamal M, Chinappa D Kodira, Todd D. Taylor et al. DNA sequence and analysis of human chromosome 18. Nature, 2005, 437: 551-555.
4. Sebold C, Soileau B, Heard P, Carter E, O’Donnell L, Hale DE, et al. Whole arm deletions of 18p: Medical and developmental effects. Am J Med Genet Part A, 2015, 167A: 313-323.
5. Turleau С. Monosomy 18p. Review Orphanet Journal of Rare Diseases, 2008, 3: 4.
6. Козлова С.И., Демикова Н.С. Наследственные синдромы и медико-генетическое консультирование. КМК, Авторская академия, Москва 2007.
7. Hasi-Zogai M, Sebold C, Heard P, Carter E, Soileau B, Hill A, et al. A Review of 18p Deletions. American Journal of Medical Genetics Part C (Seminars in Medical Genetics), 2015, 169C: 251-264.
8. Дегтярева А.В, Мухина Ю.Г, Дегтярев Д.Н. Синдром холестаза у новорожденных детей. Пособие для врачей. М: 4ТЕ АРТ, 2011. 36 с.
9. Mauvais F-X, Gonzales E, davit-Spraul A, Jacquemin E, Brauner R. Cholestasis Reveals Severe Cortisol Deficiency in Neonatal Pituitary Stalk Interruption Syndrome. PLOS ONE, 2016, February 1. DOI: 10.1371/journal.pone.0147750.
10. Braslavsky D, Keselman A, Galoppo M, Lezama C, Chiesa A, Galoppo C et. Al. Neonatal cholestasis in congenital pituitary hormone deficiency and isolated hypocortisolism: characterization of liver dysfunction and follow-up. Arq Bras Endocrinol Metab, 2011, 55/8.
11. Lammoglia J, Eyzaguirre F, Unanue N, Román R, Codner E., Cassorla F. et al. Hipopituitarismo congénito: experiencia en 23 casos. Rev Méd Chile, 2008, 136(8): 996-1006.
12. De León D, Stanely C, Sperling M. Hypoglycemia in Neonates and Infants. En: Sperling M. Pediatric Endocrinology. 3 ed. Filadelfia: Saunders. 2008: 710-5.
13. Дегтярева А.В., Пучкова А.А, Мухина Ю.Г, Лукина Л.И, Кыштымов М.В. Урсодезоксихолевая кислота в комплексной терапии неонатального холестаза. Вестник педиатрической фармакологии и нутрициологии, 2006, 3(2): 27-31.
Читайте также: