
Релаксационные свойства полимеров реферат
Обновлено: 05.07.2024
Полимер — это высокомолекулярное соединение: количество мономерных звеньев в полимере (степень полимеризации) должно быть достаточно велико (в ином случае соединение будет называться олигомером ). Во многих случаях количество звеньев может считаться достаточным, чтобы отнести молекулу к полимерам, если при добавлении очередного мономерного звена молекулярные свойства не изменяются. [1] Как правило, полимеры — вещества с молекулярной массой от нескольких тысяч до нескольких миллионов
Если образец слабо сшитого эластомера подвергнуть быстрой деформации и закрепить в растянутом состоянии, то напряжение, необходимое для поддержания этой постоянной деформации, уменьшается во времени, стремясь к некоторому постоянному значению. Этот процесс называется релаксацией напряжения и может быть представлен зависимостью либо напряжения, либо модуля упругости (G) от времени (рис. 1, кривая 1).
Релаксация – процесс структурной перестройки вещества при переходе из неравновесного состояния в равновесное, который осуществляется во времени. Большие обратимые деформации полимеров в высокоэластическом состоянии всегда развиваются во времени, поскольку они связаны с перемещением сегментов макромолекул, которые не могут произойти мгновенно. Поэтому высокоэластическая деформация является релаксационным процессом.

Рис.1 График релаксация напряжения
В общем случае модуль упругости слагается из двух составляющих: равновесного модуля (G ∞ ), зависящего от температуры и густоты химической сетки полимера, и релаксационного модуля (G t ), меняющегося во времени и обусловленного физическими узлами (флуктуационной сеткой) в полимере: G = G t + G ∞
Уменьшение релаксационного модуля происходит по экспоненциальному закону и характеризуется некоторым параметром, называемым временем релаксации. Однако зависимость релаксационного модуля от времени не подчиняется уравнению с одним временем релаксации (рис. 1, кривая 2), а может быть описана как сумма нескольких экспонент с различными временами релаксации:
Другими словами, релаксационные свойства полимеров характеризуются широким набором (спектром) времен релаксации. Это обусловлено тем, что макромолекулы не являются элементарными кинетическими единицами, а представляют собой совокупность различных по величине и подвижности кинетических единиц: боковые атомные группы, звенья цепи, сегменты, отдельные цепи, некоторые надмолекулярные образования и т. п. Подвижность каждой структурной единицы характеризуется своим временем релаксации, лежащим в интервале от малых долей секунды до многих лет.
Деформационные свойства полимерного материала зависят от соотношения времени действия силы (t) и времен релаксации (τ) его структурных элементов. Если t / τ > 1, т. е. при очень малых τ (при высоких температурах) или очень больших t, система очень быстро релаксирует и достигает равновесного состояния. Релаксационные процессы проявляются максимально при t / τ ~1, что наблюдается в высокоэластическом состоянии, особенно вблизи температуры стеклования. Для эластомеров время действия силы оказывается внутри спектра времен релаксации и для полной характеристики полимера необходимо знать его релаксационный спектр.
При достаточно больших временах действия силы (t > t’) реализуются процессы с максимальным временем релаксации (τ m ), которое может быть определено из наклона прямолинейного участка зависимости ln G t от t ln G t = ln G m - t / τ m . Из отрезка, отсекаемого на оси ординат при t = 0, определяют ln G m , где G m – доля общего модуля упругости, обусловленная подвижностью элементов со временем релаксации τ m . 3 Затем, построив зависимость ln(G t - G m ) от t, аналогично находят Gm-1 и τm-1. Повторяя эту операцию, получают набор времен релаксации и соответствующих им долей общего модуля.
От набора дискретных времен релаксации можно перейти к непрерывному спектру времен релаксации. Тогда релаксационный модуль принимает вид:

Обычно предпочитают пользоваться логарифмической шкалой времени, вводя вместо G(τ) функцию H (ln τ):
где H(ln τ) dln τ – доля модуля, которая обусловлена работой элементов с временами релаксации, лежащими в интервале от ln τ до ln τ + dln τ,
H(ln τ) – функция распределения долей общего модуля по ln τ.

Рис.4 Релаксационный спектр линейного аморфного полимера
На рис. 4 представлен типичный релаксационный спектр линейного аморфного полимера при постоянной температуре. Максимумы на спектре соответствуют временам релаксации, характеризующим подвижность различных релаксирующих структурных единиц:
γ – боковых групп,
β – мономерных звеньев (10-8 – 10-7 сек),
α – сегментов (10-6 – 10 сек)
λ1, λ2, λ3 – надмолекулярных структур, физических узлов сетки (102 – 104 сек),
δ – химических поперечных связей (107 – 109 сек).
Каждое дискретное время зависит от температуры и снижается при ее повышении:
, где Bi – постоянная, зависящая от размера структурного элемента, Ui – энергия активации перехода структурного элемента из неравновесного состояния в равновесное. В стеклообразном состоянии релаксация сопровождается γ- и β-переходами, энергии активации которых невелики и высота пиков небольшая. В высокоэластическом состоянии основную роль в релаксационных процессах играют α, λ, δ-переходы, т. е. ориентация сегментов, перегруппировки надмолекулярных структур и химических связей.
СТРУКТУРНЫЕ РЕЛАКСАЦИЯ И СТЕКЛОВАНИЕ
В каждом из релаксационных состояний — стеклообразном, высокоэластическом и вязкотекучем — могут происходить элементарные процессы, заключающиеся в перемещении определенных элементов структуры или их перестройке (включая распад на меньшие элементы). Каждому такому процессу можно в первом приближении приписать определенное время жизни т,-,

Рис. VIII. 1. Релаксационный спектр и стрелка действия
причем, как правило, чем меньше размеры структурного элемента (релаксационного структона), тем меньше его характерное т,-. Определим функцию распределения времен релаксации струк- туры д(х), которую для общности будем считать не- прерывной. Условие нормировки запишем в обычной форме:
Релаксационная концепция стеклования была впервые сформулирована одним из основоположников физики полимеров Кобеко [108]. Обычно при измерениях температуры стеклования Тст (при охлаждении) или температуры размягчения Tр (при нагревании) скорости охлаждения q = |dТ/dt| или нагревания w = dT/dt задаются в процессе опыта постоянными. Из релаксационной природы стеклования следует, что Тст и Тр с увеличением q или w должны смещаться к высоким температурам, чего никогда не наблюдается в случае фазовых переходов.
При изучении стеклования полимеров неоднократно ставился вопрос об относительной роли в нем внутри- и межмолекулярных взаимодействий. Изучение методом ИК-спектроскопии раздельного изменения внутри- и межмолекулярной энергий при стекловании показало, что при стекловании изменяются, главным образом, межмолекулярные взаимодействия; поэтому температура стеклования существенно зависит от последних. Например, атактический полибутадиен (СКВ) имеет Тст = 223 К. Если в цепь полибутадиена ввести полярные звенья крилонитрила, то межмолекулярные взаимодействия возрастают и при 50 % содержании в полимерной цепи нитрильных звеньев температура стеклования становится Тст = 263 К- Все сополимеры характеризуются тем, что с изменением концентрации сомономерных групп в макромолекуле изменяется межмолекулярное взаимодействие и изменяется Тст. В бутадиенстирольных сополимерах при переходе от атактического полибутадиена к атактическому полистиролу Тст изменяется от 223 до 373 К.
Практические применения в качестве стекол или конструкционных пластиков имеют полимеры с молекулярной массой, заведомо большей критической. Поэтому в дальнейшем мы будем под Тст понимать температуру стеклования собственно полимеров, не зависящую от М.
Структурное стеклование полимеров связано с потерей кооперативной межцепной сегментальной подвижности при Тст и переходом некристаллического полимера или аморфной фазы кристаллического полимера из высокоэластического (структурно-жидкого) состояния в стеклообразное. Процесс сегментальной релаксации, протекающий при переходе полимера из неравновесного состояния в равновесное, моет наблюдаться в различных условиях опыта. Если система приведена в неравновесное структурное состояние в отсутствие внешних силовых воздействий, то наблюдается структурная релаксация, сопровождающаяся изменением функций состояния.
ВЯЗКОУПРУГИЕ СВОЙСТВА ПОЛИМЕРОВ
И ПРОЦЕССЫ МЕХАНИЧЕСКОЙ РЕЛАКСАЦИИ
В реальных твердых телах, в том числе полимерах, упругая энергия при деформациях рассеивается (существует внутреннее трение). В результате, если приложено постоянное или периодическое напряжение, то деформация отстает по фазе от напряжения, тогда как для идеально упругих твердых тел это явление должно отсутствовать. Если задается деформация, то напряжение опережает по фазе деформацию, а при постоянной степени деформации ε = const наблюдается релаксация напряжения σ = σ(t). Если твердое тело подчиняется закону линейной упругости, то для него верен закон Гука. В случае растяжения:
В более общем случае нелинейной упругости закон деформации усложняется:
Если напряжение превышает предел упругости, то его удобнее представить в виде:
При больших деформациях, когда закон упругости нелинеен, но структура материала не изменяется, наблюдается геометрическая или деформационная вязкоупругость:
Такой закон соблюдается для сшитых эластомеров.
Принцип суперпозиции Больцмана применим для всех полимеров, структура которых не зависит от приложенных сил и ие меняется во времени. Ои позволяет описывать линейное вязкоупругое поведение системой дифференциальных уравнений вида: Lσ = Dε, где L и D—линейные дифференциальные операторы по времени. Это выражение эквивалентно описанию вязкоупругого поведения с помощью моделей, состоящих из упругих пружин с различными модулями Ei и вязких элементов с вязкостями ηi (рис. IX. 2). Пружинам приписываются механические свойства идеальной упругости — закон Гука, а вязким элементам — свойства идеально вязкой жидкости — закон Ньютона.

Модели Максвелла и Кельвина — Фойгта
Самая простая модель упруговязкого материала — модель Максвелла (рис. IX. 2, а). Пусть упругий элемент (пружина) характеризуется деформацией ε1 и напряжением σ1 а вязкий элемент — ε 2 и σ2. Очевидно, что растягивающая сила и напряжение при одном и том же поперечном сечении одинаковы вдоль действия сил, поэтому σ1 = σ2 =σ. Упругий элемент подчиняется закону Гука, а вязкий — закону Ньютона, поэтому:
Отсюда, учитывая, что
![]()
Для релаксации напряжения (ε= const ) получим dσ / dt = -( E / η ) σ ; интегрируя от 0 до t и от σ о до σ, придем к известному закону релаксации Максвелла (рис. IX . 3, а):
![]()
где τ = η/Е — время релаксации (константа материала при Т = const ,
имеющая размерность времени).
Этот закон качественно верен для вязких материалов, обла-
дающих упругостью (упруговязкие тела). Для твердых тел с
внутренним трением (вязкоупругие тела) модель Максвелла не

описывает ползучесть. Простейшая модель для вязкоупругих тел, которые не обнаруживают вязкого течения, это модель Кельвина — Фойгта (рис. IX . 2, б). Соответствующее линейное дифференциальное уравнение имеет вид:
![]()
Для ползучести (σ = const) получим уравнение:
![]()
где ε х = σ/Е; τ n = η n /E — время ползучести (константа, аналогичная вре-
мени релаксации); η n — вязкость ползучести (константа, характеризующая
внутреннее трение в полимерах).
![]()
где в скобках записан комплексный модуль E ф = Е' + Е", поэтому Е' = Е;
Е" = ωη и tg δ = ωη п /Е.
Рассмотренные простейшие модели даже качественно не описывают основные вязкоупругие свойства. Так, модель Максвелла не описывает ползучесть, а модель Кельвина — Фойгта — релаксацию напряжения. Простейшая модель, качественно описывающая основные вязкоупругие свойства, — это модель стандартного линейного тела , называемая также моделью Зинера.
Обобщенная модель Максвелла и дискретные формы молекулярного движения в полимерах
Тобольский [143] и другие исследователи применяли обобщенную модель Максвелла (см. рис. IX. 2, д) чисто феноменологически: чем больше констант, тем лучше описываются особенности вязкоупругих свойств полимеров. Между тем, дискретность строения полимеров и существование в них многоуровневой надмолекулярной организации позволяют выделить реальные дискретные релаксационные процессы, число которых связано с числом уровней организации (подсистем). Поэтому физически оправдано применение обобщенной модели Максвелла, представленной на рис. IX. 5, где А, В, С. . М — различные подсистемы полимера. Дискретность спектра оправдана, если времена релаксации тА, тв и т. д. отстоят друг от друга достаточно далеко, чтобы каждый релаксационный переход был четко

разрешен на спектре. Для многих полимеров это условие соблюдается.
ПРОЦЕСС α-РЕЛАКСАЦИИ И МЕХАНИЧЕСКОЕ СТЕКЛОВАНИЕ
Впервые Александровым и Лазуркиным [107] в 1939 г. убедительно было показано, что переход из стеклообразного в высокоэластическое состояние есть релаксационный процесс. В дальнейшем его назвали α -процессом. Эти же авторы показали, что с увеличением частоты деформации область перехода сдвигается к более высоким температурам.
Как потом оказалось, температуры структурного стеклования Тст и механического стеклования Т α отличаются, хотя молекулярная природа структурного и механического стеклования одна и та же и связана с сегментальным движением. Причины различия между обоими видами стеклования были рассмотрены в работе автора. Там же для периодических деформаций был введен термин механическое стеклование.

Природа механического стеклования — перехода жидкости (полимера) из вязкого (высокоэластического) состояния в упругое, и природа структурного
стеклования одна и та же и определяется одними и теми же процессами молекулярных перегруппировок. Однако при механическом стекловании переход в упруготвердое состояние не связан с замораживанием структуры и происходит в структурно-жидком состоянии системы, т. е. выше температуры структурного стеклования.
Линейная теория вязкоупругости является математической основой для описания релаксационных свойств полимеров.
Для качественного описания вязкоупругости полимеров применяются различные механические модели: Максвелла, Кельвина — Фойгта, обобщенные модели Максвелла и др.
Среди релаксационных процессов важнейшим для полимеров является а-релаксация (стеклование). При этом в зависимости от того, действуют на полимер внешние силы или нет, наблюдается механическое или структурное стеклование, зависящие соответственно от частоты и скорости охлаждения.
Ниже температуры структурного стеклования Тст механическое стеклование не наблюдается. Структурная и механическая релаксация являются наиболее универсальными методами исследования релаксационных переходов в полимерах и важно, что имеется определенная взаимосвязь между механическими и структурными релаксационными переходами.
Акустическая спектроскопия, как метод исследования высокочастотных релаксационных переходов, занимает особое место среди других методов механической релаксации, особенно при исследовании вязкоупругих свойств растворов полимеров.
В общем случае под релаксационными явлениями в полимерах понимают изменение их свойств во времени, обусловленное достижением равновесного состояния. В принципе релаксационные явления должны иметь место при любых процессах, протекающих в полимерных системах, связанных с подвижностью макромолекул или ее фрагментов. Известны электрические, магнитные, механические релаксационные явления, явления, наблюдаемые при плавлении, кристаллизации, стекловании, растворении, набухании полимеров. При изучении свойств полимеров в широком интервале температур проявляются так называемые релаксационные переходы, связанные с возникновением или исчезновением подвижности тех или иных фрагментов макромолекул или макромолекулы в целом.
В основе механических релаксационных явлений в полимерах, которые проявляются как зависимость упругости и вязкости от времени, лежит явление вязкоупругости. Выше с использованием моделей Максвелла и Кельвина были охарактеризованы важнейшие вязкоупругие свойства полимеров:
релаксация напряжения и ползучесть при статическом характере нагружения;
отставание деформации от напряжения (или наоборот) при динамических нагрузках или деформациях.
Для описания релаксационных свойств реального полимера, т.е. зависимости тех или иных его свойств от времени, необходимо:
а)использовать обобщенную модель (см. рис. 2.28, в) для того, чтобы отразить все возможные виды деформаций;
б)увеличить количество вязких и упругих элементов в модели с различными Е и ŋ пружины и демпферов. При этом будет получен спектр времен релаксации и времен запаздывания θ, позволяющий со сколь угодно большой точностью приблизиться к количественному описанию релаксационных свойств полимеров.
На рис. 2.34 приведена зависимость, отражающая развитие деформации во времени обобщенной модели вязкоэластоупругого полимерного тела.
Как видно из рис. 2.34, при приложении напряжения σ0 = const начинает развиваться деформация, включающая три составляющие:
εупр - обратимая упругая деформация, развивающаяся мгновенно, обусловлена деформацией упругого элемента - пружины с модулем E1 последовательно присоединенной к модели, она равна σ0/E1. В полимере развивается за счет незначительного изменения значений углов и длины связей;
εэл - высокоэластичная обратимая деформация, обусловленная растяжением пружины с модулем Е2 и демпфера, связанных параллельно, равна
ε0/Е. В полимерном теле реализуется за счет подвижности сегментов;
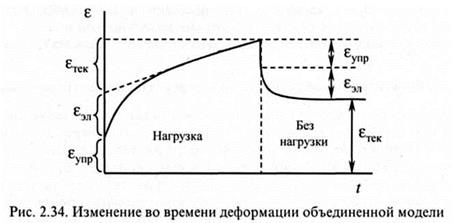
εтек - необратимая деформация течения, развивается за счет растяжения демпфера, присоединенного к модели последовательно, в полимерном теле -за счет перемещения макромолекул.
После сброса нагрузки мгновенно исчезает εупр, затем в течение некоторого времени исчезает εэл, εтек - необратима.
На рис. 2.35 представлены кривые изменения относительной деформации нагруженного полипропилена во времени. Такие кривые обычно называют кривыми ползучести, однако, это не совсем корректно, ибо к истинной ползучести относят лишь ту, которая связана с эластичностью (см. разд. 2.3.3).
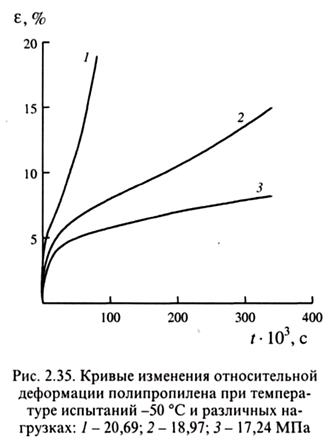
Можно сделать вывод, что:
1. εупр достигает 3-5 %;
2. εэл развивается за время, порядка минут, в этом случае эластичность характеризуется модулем эластичности ;
Из рис. 2.35 видно, что в области установившейся ползучести деформация возрастает быстрее, чем это следует из закона Ньютона.
Принцип температурно-временной суперпозиции. Результаты динамических испытаний полимеров показывают, что влияние температуры и частоты на отставание деформации от напряже ния эквивалентно. Из рис. 2.36
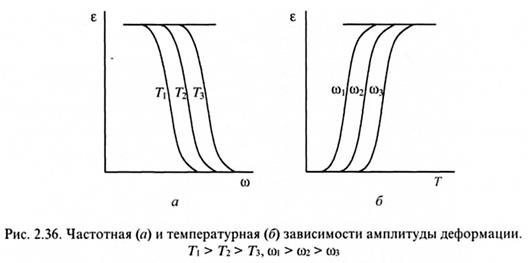
следует, что при больших частотах и низких температурах деформация не успевает развиваться из-за малого времени воздействия и малой подвижности сегментов макромолекул. При малых частотах и высоких температурах деформация достигает максимально возможного (равновесного) значения, так как в этих условиях подвижность сегментов реализуется полностью.
Отставание деформации от напряжения в этих опытах обусловлено тем, что время релаксации, понимаемое как время перестройки полимера, отстает от времени воздействия порядка периода колебаний. Однако это время можно существенно сократить путем повышения температуры, т.к. при этом происходит увеличение интенсивности различных форм молекулярного движения. В общем случае:

где В - постоянная, зависящая от объема структурного элемента. Следовательно, глубина релаксации определяется двумя факторами - временем и температурой и их влияние эквивалентно. Эквивалентность факторов времени и температуры позволяет осуществлять взаимный пересчет вязкоупругих характеристик полимерных материалов, такой подход носит название принципа температурно-временного приведения или температурно-временной суперпозиции.
Читайте также: