
Реферат на тему онкогены
Обновлено: 04.07.2024
За последние 25 лет произошел существенный прогресс в нашем понимании молекулярных механизмов развития злокачественных опухолей. Идентифицировано три типа генов, нарушения в которых приводят к раку: доминантные трансформирующие, или онкогены; рецессивные трансформирующие, или опухолевые супрессоры; гены, ответственные за репарацию ДНК. Многие онкогены были впервые выделены как формы протоонкогенов онкогенных РНК-содержащих вирусов.
На протяжении многих лет известно, что вирусы способны вызывать злокачественные опухоли у животных. Это наблюдение послужило стимулом к обширным исследованиям, направленным на выявление генов, которые вызывают рак и переносятся вирусами, и генов человека, повреждающихся при развитии злокачественных опухолей. В итоге обнаружили удивительный факт, что гены, вовлеченные в канцерогенез, часто представляют собой измененные формы вирусных генов.
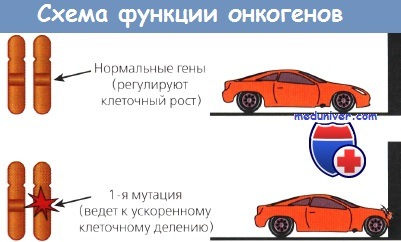
Другие протоонкогены кодируют внутриклеточные белки, ответственные за усиление митогенного сигнала; третьи — кодируют белки, вовлеченные в контроль клеточного деления и находящиеся под контролем ядра. Такие онкогены могут быть активированы посредством нескольких механизмов: возможна амплификация гена и активация его копий; в редких случаях наблюдается траислокация гена на другую хромосому, где он попадает под контроль чужого промотора и стимулирует неконтролируемый рост. Три группы онкогенов находятся под разным контролем.
Первая группа включает пептидные факторы роста и их рецепторы, как, например, эпидермальный или тромбоцитарный.
Эти пептиды служат скорее костимуляторами опухолевого роста, чем факторами, непосредственно инициирующими опухолевую трансформацию. По мере расшифровки механизмов, стимулирующих опухолевую прогрессию, все более реальной становится молекулярно-нацеленная (таргетная) терапия, направленная непосредственно против продуктов этих генов или против белков, активируемых ростовыми факторами. Так разработаны препараты:
1) трастузумаб (Герцептин) — моноклональные антитела, блокирующие Her-2/neu;
2) иматиниба мезилат (Гливек) — препарат, частично блокирующий активность c-kit, BCR-ABL и некоторых других тирозинкиназ;
3) цетуксимаб (Эрбитукс) — моноклональные антитела, связывающиеся с рецептором эпидермального фактора роста;
4) гефитиниб (Пресса) — низкомолекулярный ингибитор некоторых изоформ рецептора эпидермалыюго фактора роста.

Другой класс онкогенов происходит из немембранных внеядерных факторов роста, передающих каскадные сигналы внутри клетки. К ним относятся G-белки и белки семейства ras. Наконец, некоторые онкогены кодируют ядерные регуляторные белки, например myc. Схематическое изображение функций онкогенов представлено на рисунке.
Открытие человеческих аналогов вирусных генов привело к формулировке многообещающей гипотезы, согласно которой злокачественные опухоли человека, включая большинство невирусной этиологии, могут возникать вследствие мутаций, превращающих полезные протоонкогены в опасные онкогены. В подтверждение этой гипотезы показано, что повреждение даже одного аллеля таких протоонкогенов достаточно для злокачественной трансформации некоторых клеток in vitro. Подобные доминантные мутации ведут к гиперэкспрессии нормального гена и, следовательно, гиперпродукции нормального белка или же к синтезу его аберрантной формы, обладающей повышенной активностью.
В любом случае результатом оказывается усиление стимулирующих сигналов внутри клетки, даже в отсутствие стимулирующих воздействий извне.
Парадоксально, но первым ключом к выявлению некоторых онкогенов стало исследование РНК-содержащих онкогенных вирусов животных, не вызывающих опухолей у человека. Эти ретровирусы, инфицирующие кур, грызунов, кошек и обезьян, оказались крайне высокоонкогенными: развитие опухоли часто наблюдали уже при первом контакте. У одного из вирусов этой группы, вируса саркомы кур Рауса, выявлен ген, ответственный за злокачественное перерождение инфицированных клеток. Этот тип трансформирующих онкогенов был назван вирусным онкогеном.
Единственный онкоген вируса саркомы Рауса, проникая в клетки кур, способен нарушать и перестраивать их метаболизм, направляя его по пути злокачественной трансформации.
В 1976 г. Varmus и Bishop показали, что онкоген вируса саркомы Рауса в действительности вовсе не есть вирусный ген, а происходит от предсуществовавшего клеточного гена, захваченного предком вируса саркомы Рауса. Однажды встроив его в свой геном, вирус далее использовал этот ген для трансформации клеток млекопитающих.
Более ранние предки вируса саркомы Рауса были способны реплицироваться в инфицированных клетках, но не могли трансформировать ее; туморогенный потенциал был приобретен после захвата нормального клеточного гена — протоонкогена. В связи с этим значение работы Varmus и Bishop гораздо больше, чем просто исследование вируса саркомы Рауса: показано существование гена в нормальном геноме клеток млекопитающих, обладающем трансформирующим потенциалом при соответствующей активации, в данном случае ретровирусом.
Информация, полученная при исследовании ретровирусов и онкогенов, существенно помогла в изучении причин злокачественных новообразований. Ретровирусы, так же как вирус саркомы Рауса, неинфекционны для человека и, соответственно, не могут активировать протоонкогены человека. Однако возможны альтернативные механизмы их активации. Эффекты, сходные с производимыми вирусом в последовательности ДНК, могут быть вызваны химическими или физическими воздействиями.
Это было подтверждено в начале 80-х годов прошлого столетия: в геноме опухолевых клеток были выявлены мутированные гены (протоонкогены). Во всех случаях причиной превращения протоонкогена в активный онкоген оказались изменения последовательности гена. Например, онкоген ras образуется из протоонкогена-предшественника в клетках рака мочевого пузыря человека вследствие замены одной пары нуклеотидных оснований; онкоген myc появляется во многих злокачественных новообразованиях в результате амплификации.
В последующем были изучены механизмы, посредством которых большинство, если не все онкогены вызывают трансформацию клеток. Это стало возможным благодаря определению путей, посредством которых клетки регулируют свой собственный рост. Рост иделение нормальной клетки вткани контролируется преимущественно ее окружением. В норме клетка редко или даже никогда не определяет скорости своего деления, а только реагирует на сигналы от окружающих клеток.
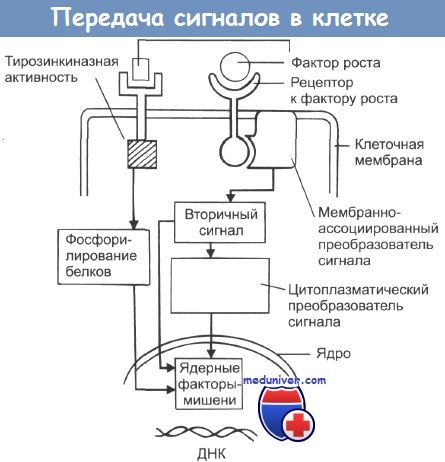
Эти сигналы, стимулирующие или подавляющие рост, передаются посредством ростовых факторов, выделяемых окружающими клетками. Ростовые факторы попадают в межклеточное пространство и связываются с рецепторами на поверхности клеток-мишеней. Клетки реагируют на сигналы ростовых факторов активацией механизмов синтеза клеточных структур, удвоением ДНК и делением. Нормальная клетка никогда не запускает программу роста, если она не получила внешнего сигнала. Каждая клетка обладает сложной системой, позволяющей ей получать ростовые сигналы, обрабатывать их и запускать программу деления. Эта система состоит из большого количества белков, ответственных за получение ростовых сигналов и передачу их в клетку. К этим белкам относятся:
1) рецепторы на клеточной поверхности, распознающие наличие во внеклеточном пространстве ростовых факторов и передающие сигнал во внутриклеточное пространство;
2) белки внутриклеточной сигнальной системы, которые активируются поверхностными рецепторами и затем передают сигнал дальше в клетку;
3) ядерные транскрипционные факторы, активирующиеся в ответ на сигнал, переданный белками внутриклеточной сигнальной системы, и, в свою очередь, активирующие широкий спектр клеточных генов.
Активирующиеся гены руководят программой роста клетки; именно эти гены определяют те события, которые вместе взятые приведут к делению клетки. Протоонкогены кодируют многие белки в этой сложной сигнальной системе, позволяющей нормальной клетке отвечать на экзогенные ростовые факторы. Белки онкогенов участвуют в сигнальной системе, отбирая ее неправильно функционирующие версии нормальных компонентов, и вызывают ее постоянную стимуляцию в отсутствие внешних ростовых сигналов. В результате клетка постоянно растет, даже если окружающая среда не содержит каких-либо факторов, в норме необходимых для клеточного роста.
Некоторые исследователи указывают, что в злокачественной клетке должны присутствовать как минимум две мутации в протоонкогенах, причем только строго определенные мутации могут вызывать злокачественную трансформацию. Это связано с тем, что отдельные онкогены, даже если они служат важными регуляторами клеточного метаболизма, сами по себе не способны индуцировать злокачественное новообразование. Данная точка зрения подтверждается выявлением в опухолевых клетках более 10 различных онкогенов. Однако при тщательном исследовании ожидаемые нарушения обнаруживают лишь примерно в 20 % опухолей.
Ни одна опухоль не несет ни одной пары сочетанных нарушений из выявляемых в культивируемых линиях злокачественных клеток. Предполагается также, что врожденные мутации, ответственные за предрасположенность людей к раку, не есть онкогены. Это объясняется существованием рецессивных антионкогенов, получивших название генов-супрессоров, тоже играющих крайне важную роль в развитии опухолей.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Студент ББ14-04Б ____________ _________ Сковородина Ю.А.
Красноярск 2018
Содержание
Используемая литература: 4
Основная часть 5
1.Стадии и причины малигнизации клеток 5
2.Свойства приобретенные нормальной клеткой в процессе малигнизации 7
Инициация канцерогенеза происходит за счет мутаций в генах, регулирующих клеточное деление — протоонкогенах и генах-супрессорах (антионкогенах) [4]. 7
3. Понятие об онкогенах и протоонкогенах 7
Онкогены — это участки ДНК (гены), функционирование которых приводит к опухолевой трансформации клеток. Открытие онкогенов имело большое значение для исследования фундаментальных механизмов канцерогенеза. Впервые онкогены были найдены в опухолеродных вирусах и идентифицированы как факторы, ответственные за процесс трансформации (вирусные онкогены) [2]. 7
4.Механизм перехода протоонкогена в онкоген 8
5.Стадии канцерогенеза 11
Введение
По статистике в мире ежегодно выявляют более 6 млн. случаев заболеваний раком пяти основных органов (легких, желудка, груди, прямой кишки, шейки матки). Около половины заболевших людей погибает. В конечном счете, каждый пятый житель развитых стран умирает от онкологических заболеваний. Это само по себе говорит об исключительной важности онкологических исследований даже в чисто прикладных целях.
Исследования изменений в функционировании клеток, возникающих при злокачественном перерождении, имеют фундаментальное теоретическое значение.
Наиболее интенсивные исследования биологии клетки и медицины касаются процесса образования и формирования злокачественных опухолей – канцерогенез, который имеет генетическую природу.
Онкогенетика- раздел онкологии, изучающий роль генетических факторов в этиологии и патогенезе опухолей. Различные физические, химические и биологические (вирусные) факторы могут привести к злокачественной (неопластической) трансформации клетки – малигнизации.
Изучить основы онкогенетики, трансформации клеток в процессе опухолеобразования, онкогены.
1) Ознакомиться с основами онкогенетики
2)Выявить причины и виды трансформаций клеток в процессе опухолеобразования
3) Изучить типы онкогенов и их влияние на онкологию
4)Сделать краткий обзор по полученной информации
Используемая литература:
1.Горбунова В.Н., Имятников Е.Н. Генетика и канцерогенез. Федеральное агентство по здравоохранению и социальному развитию: Методическое пособие. - С.-Петербург, 2007. - 24 с.
2. Жимулев И.Ф. Общая и молекулярная генетика. - Новосибирск: Сибирское университетское издательство, 1998. - 430 с.
3.Сальникова Л.Е. Генетическая детерминация эффектов ионизирующих излучений: цитогенетические и эпидемиологические показатели. // Диссертация на соискание ученой степени доктора биологических наук. - Москва, 2011. - 320 с.
Выполнил: студент 110 группы
Стоматологического факультетаРужьин И.Н.
Проверила: Якушкина Е.В.
Смоленск 2014
Оглавление:
1. Онкоген
2. Протоонкогены
3. Онкогенные вирусы
4. Трансформацияонкогенов в опухолевые клетки
5. Список использованной литературы
Онкоген
Онкоген — это ген, кодирующий белок, который, в случае нарушения регуляции, может вызвать образование злокачественной опухоли. Мутации, вызывающие активацию онкогенов, повышают шанс того, что клетка превратится в раковую клетку. Первый онкоген, названный src , был выделен из вируса куриной саркомы. Вскоребыло показано, что искусственное введение гена src в генетический аппарат клетки трансформирует ее без вируса. Один-единственный ген, введенный в клетку, превращал ее в опухолевую! Вскоре после открытия src-гена были открыты и другие вирусные онкогены: myc , ras , abl и многие другие
Онкогены обусловливают превращение нормальных клеток эукариот на злокачественные при участии онкобелков,которые они кодируют. Образуются онкогены с видоизмененных нормальных генов (проонкогенов), которые широко представлены в разных видах организмов. Образование проонкогенов происходит вследствие точечных мутаций, амплификации и усиления экспрессии генов, хромосомных перестроек.
Известно около 30 онкогенов, которые кодируют соответствующие белки. В злокачественном перерождении клеток принимаютучастие, как правило, два онкогена. Значительное количество онкобелков характеризуется протеинкиназной активностью, специфической для аминокислоты тирозина. Фосфорилирование тирозина является одним из пусковых моментов каскада злокачественного перерождения клеток.
Считается, что гены-супрессоры опухолей (ГСО) предохраняют клетки от ракового перерождения, и, таким образом, рак возникает либо в случаенарушения работы генов-супрессоров опухолей, либо при появлении онкогенов.
Многие клетки при появлении в них мутаций вступают в апоптоз, но в присутствии активного онкогена могут ошибочно выживать и пролиферировать. Для злокачественного перерождения клетки под действием многих онкогенов требуется дополнительная стадия, например, мутация в другом гене, факторы внешней среды (например,вирусные инфекции).
С 1970х открыты десятки онкогенов, вызывающие канцерогенез у человека, многие противораковые лекарства направлены на подавление активности онкогенов либо их продуктов.
Множество генов было идентифицировано как протоонкогены( Альбертс Б., 1994). Многие из этих генов ответственны за передачу сигналов, ведущих к клеточному делению. Некоторые изпротоонкогенов ответственны за клеточную гибель. Как было сказано во введении к этому разделу, дефектные версии этих генов, называемые онкогенами, могут привести к нерегулируемому клеточному делению. Такое деление может происходить в отсутствии нормальных ростовых сигналов, обеспечиваемых факторами роста. Основным признаком активности онкогенов является то, что изменения лишь в одной аллели протоонкогенаведут к нерегулируемому росту. Этим они отличаются от генов-супрессоров опухолевого роста, в случае которых к аномальному клеточному делению ведёт дефект ОБЕИХ аллелей.
Протоонкогены, выявленные к настоящему времени, выполняют в клетке множество различных функций. Несмотря на различия их нормальных функций, все эти гены вносят свой вклад в нерегулируемое клеточное.

Представления о молекулярно-клеточных механизмах онкогенной трансформации клеток претерпели значительную эволюцию на протяжении XX века и до настоящего времени [18, 20, 25, 32, 34].
Как указывалось выше, инициирующими этиологическими факторами малигнизации клетки являются разнообразные по природе группы канцерогенов химической, физической, биологической природы, в том числе вирусы, гормоны и генотоксические продукты их метаболизма [13, 26, 63].
Естественно, что при чрезвычайной гетерогенности этиологических факторов неоплазий не могла сформироваться достаточно быстро доминирующая концепция механизмов развития онкогенной трансформации клеток, их активации или промоции опухолевого роста с последующей опухолевой прогрессией. В ранних концепциях канцерогенеза делали акцент на эпигеномных механизмах развития неоплазий, и, безусловно, ряд положений этого направления носит не только исторический характер, но может быть в определенной степени ассоциирован с современными вирусо-генетической и онкогенной теориями канцерогенеза.
Согласно данным ряда исследователей, первичное изменение свойств цитоплазматической мембраны под влиянием канцерогенных углеводородов, онкогенных вирусов является одним из пусковых механизмов последующего изменения генетического аппарата и нарушений регуляции их митотического цикла [108]. Эта концепция канцерогенеза была актуальна в период обнаружения отсутствия контактного ингибирования опухолевых клеток в монослойной культуре.
Как оказалось далее, в механизмах контактного ингибирования клеток важная роль отводится активации мембранной аденилциклазы и увеличению уровня цАМФ, тормозящего митотическую активность клеток. Понижение концентрации цАМФ в мембранах клеток под влиянием различных канцерогенов ведет к неконтролируемой митотической активности. Эта точка зрения имела определенную значимость в понимании пусковых механизмов канцерогенеза, поскольку для многих гормонов, регулирующих метаболизм клеток, их митотическую активность, характерен преимущественно мембранный тип рецепции (АКТГ, СТГ, инсулин, пролактин и др.).
Практически одновременно с мембранной концепцией канцерогенеза создавались митохондриальная и лизосомальная теории развития неоплазий, согласно которым актомиозиновый белок митохондриальных мембран оказывается аномальным у малигнизированных клеток и утрачивает чувствительность к регулирующим влияниям АТФ; при этом гликолитическая реакция опухолевой клетки стимулируется митохондриальными факторами, поступающими постоянно в гиалоплазму, а возрастание концентрации АТФ не подавляет этот процесс.
Одним из классических признаков неоплазий является нарушение регуляции дифференцировки и митотической активности клеток, в связи с чем указанная проблема затрагивается в той или иной форме в разных концепциях [1]. Однако до настоящего времени одной из ведущих концепций канцерогенеза является мутационная теория, согласно которой все канцерогены обладают мутагенной активностью, хотя не все мутагены являются канцерогенами.
Практически все изученные канцерогены индуцируют разрывы фосфодиэстеразных связей в молекуле ДНК. Вначале канцерогены интенсивно связываются с ДНК чувствительных клеток. Обнаружена прямая корреляция между чувствительностью животных и их органов к малигнизирующему действию веществ и степенью их связывания с ДНК [42].
В последующие годы важная роль в развитии онкогенной трансформации клеток и опухолевой прогрессии отведена свободным радикалам. Учитывая значимость индукции избыточных концентраций свободных радикалов в канцерогенезе, необходимо прежде всего остановиться на активации процессов липопероксидации, инициируемой активными формами кислорода (АФК) и в то же время являющейся источником образования значительного количества вторичных эндогенных свободных радикалов [7, 8].
Как известно, активные формы кислорода вступают во взаимодействие с полиненасыщенными жирными кислотами (ПНЖК): линолиевой, линоленовой, арахидоновой – важнейшими компонентами фосфолипидов биологических мембран. Отрыв водорода от молекулы ПНЖК при участии АФК приводит к перемещению двойных связей с образованием гидроперекисей диеновых коньюгатов, которые затем метаболизируются во вторичные (малоновый диальдегид) и третичные продукты липопероксидации [66]. Перекисное окисление липидов затрагивает прежде всего фосфолипиды цитоплазматических мембран клеток, нарушая при этом энергозависимый трансмембранный перенос субстратов, процессы межклеточного взаимодействия. Биологическая активность АФК связана с синтезом простагландинов, лейкотриенов окислительной модификацией белков, нуклеиновых кислот, липидов. Одним из проявлений окислительной модификации белка является инактивация около 240 ферментов, в частности, СОД, ацетил-КоА-гидролазы, каталазы, миелопероксидазы, цитохрома Р450 [22, 66].
Дезинтеграция белка в основном возникает под влиянием гидроксильного радикала, образующегося в организме в процессе реакции взаимодействия супероксида и перекиси водорода с металлами переменной валентности. Объектами окисления в молекуле ДНК под влиянием гидроксильного радикала являются углеводные компоненты, фосфатные группировки, азотистые основания. Наиболее чувствительным к окислительной деструкции азотистым основанием является гуанин, модифицированные формы которого составляют 45 % от общего количества окисленных оснований [83, 95].
Установлено, что чувствительность к фрагментации сахарно-фосфатного остатка ДНК под влиянием АФК оказалось более высокой, чем полипептидного остова белково-пептидных субстанций. Гидроксильный радикал, действуя на ДНК, может отрывать атом водорода от дезоксирибозофосфата, что ведет к его расщеплению и освобождению азотистых оснований. При этом образуются высокотоксичные производные альдегиды.
Данные, опубликованные в последние годы, убедительно свидетельствуют о том, что активные формы кислорода, оксид азота и его производные в сочетании с инфекционными патогенными факторами, бактериями и вирусами, являются ключевыми факторами канцерогенеза [2, 35, 36].
Детальный обзор литературы по этому вопросу приведен в работе Х. Маеда, Т. Акаике (1998). Кислородные радикалы, а также оксид азота могут повреждать ДНК, вызывая мутацию. Мутагенный и канцерогенный эффекты указанных соединений резко возрастают при одномоментной, избыточной продукции, сопровождающейся их взаимодействием с образованием пероксинитрита. Последний участвует в различных внутриклеточных метаболических процессах: нитровании остатков тирозина в белках, подавлении транспорта электронов в митохондриях, в окислении тиоловых соединений. Пероксинитрит является ДНК-расщепляющим агентом. Вышеуказанные химические реакции с участием пероксинитрита могут инициировать апоптоз, мутации, онкогенную трансформацию клеток.
Как указывалось выше, в механизмах индукции канцерогенеза важная роль отводится онкогенным ДНК- и РНК-содержащим вирусам, способным инкорпорировать свою ДНК или ДНК-копию в геном хозяина с последующей возможной онкогенной трансформацией клетки в случае экспрессии протоонкогенов.
Установлено, что РНК-содержащие онкогенные вирусы являются членами семейства ретровирусов, характеризуются наличием липидной оболочки и двух односпиральных РНК, фермента РНК-зависимой ДНК-полимеразы, необходимой для репродукции вируса. Наличие этого фермента обеспечивает обратную транскрипцию вирусной РНК- в ДНК-копию, интегрирующую с геномом клетки [71].
Группа РНК-содержащих вирусов включает следующие разновидности: непатогенную для человека группу вирусов (род А); медленно трансформирующийся вирус гормонзависимой карциномы молочной железы морских свинок и, возможно, человека (род В); дефектные быстро трансформирующиеся и недефектные медленно трансформирующиеся вирусы (род С); род Д – включает вирусы приматов и вирус перевиваемых раковых клеток человека.
ДНК-содержащие онкогенные вирусы подразделяются на следующие семейства:
1. Семейство Poxviridae, содержит, в частности, вирус контагиозного моллюска человека.
2. Семейство Herpes viridae, к которому относится вирус Эпштейн-Барра человеа, вызывающий лимфому Беркитта, цитомегаловирус человека – тип 5.
3. Семейство Adenoviridae – представителями которого являются аденовирусы человека.
4. Семейство Papovaviridae, представителями которого являются вирусы папилломы крыс, хомяков, обезьян, человека.
ДНК-содержащие вирусы внедряют свою ДНК в геном хозяина при участии ферментов эндонуклеаз и липаз, а за счет наличия генов – промоторов – вирусы инициируют транскрипцию генов, следующих за ДНК-вирусами. Последствия внедрения ДНК-вирусов в геном хозяина зависят от зоны инкорнации: интронов, экзонов, протоонкогенов, антионкогенов. Если ДНК-содержащие вирусы встраивают в геном хозяина клетки регуляторы экспрессии протоонкогенов, возможна малигнизация клетки [54].
Механизмы онкогенной трансформации клеток под влиянием ДНК-содержащих вирусов могут быть весьма разнообразны: за счет индукции ранних онкобелков, так называемых Т-антигенов, усиления экспрессии рецепторов экзогенных ростовых факторов. Большие и средние Т-белки ряда ДНК-содержащих вирусов выключают контактное ингибирование пролиферации клеток, препятствуют действию антионкогена р53.
Как известно, вирусо-генетическая теория Л.А. Зильбера явилась основной для формирования современной онкогенной теории канцерогенеза. На смену вирусогенетической теории канцерогенеза пришли теории онкогенов, протоонкогенов и антионкогенов [30, 31, 65, 120].
В настоящее время, очевидно, что в опухолевой трансформации клеток, возникающей под влиянием различных индукторов канцерогенеза, принципиально участвуют следующие категории генов:
1. Онкогены- стимуляторы функций.
2. Гены роста и пролиферации клеток (Myc, Ras, Los, ABL и другие).
3. Антионкогены (потеря функции).
4. Гены, отвечающие за программированную смерть клетки (апоптоз):
– отменяющие программированную смерть: Bcl-2 (стимуляция функций);
– гены смерти клеток – р53 (потеря функции).
Онкогены как специфический химический материал, кодирующий информацию об определенном химическом продукте, впервые были идентифицированы в составе ретровирусов. Геном типичного не трансформирующего ретровируса представляет собой две молекулы односпиральной РНК. Основные гены вируса относятся к трем регионам: gag кодирует структурные белки вирион частицы, env– белки оболочки вириона, ген pol – несет информацию об обратной транскрипции. Последний обеспечивает образование ДНК- копии на матрице РНК-вируса.
Согласно гипотезе онкогенов, гены ретровирусов, попавшие в геном человека в процессе эволюции, переходят по наследству в ряде поколений, проявляют себя в раннем онтогенезе, а затем подавляются внутриклеточными репрессорами. С возрастом под влиянием различных канцерогенов физической, химической, биологической природы возникают экспрессия вирусных онкогенов и усиление продукции ими онкобелков, ответственных за малигнизацию клетки. Онкогенные свойства нетрансформирующих ретровирусов обусловлены наличием в их геноме V-онкогенов, причем большинство из 50 V-онкогенов имеют клеточные прототипы – С-протоонкогены.
Высказывается мысль, что ретровирусы не только могут вносить в определенные позиции клеточного генома V-онкогены, но и способны быть промоторами для усиленной экспрессии протоонкогенов клеток. Считается, что в ходе совместной эволюции ретровирусов и клеток происходят захват клеточных протонкогенов вирусами и их перенос [24].
Развитие теории онкогенов нашло отражение в концепции Темина (1972) о протовирусах, протоонкогенах, согласно которой предсуществующий аналог вируса не является результатом инфекции, а нормальным клеточным геном, необходимым для роста и онтогенеза клеток, причем нормальные клетки не содержат вирусных онкогенов, но зависят от контролируемой экспрессии их клеточных аналогов.
В механизмах развития неоплазий онкогенные ретровирусы играют неоднозначную роль: различают быстро- и медленно-трансформирующие вирусы. Быстротрансформирующие вирусы дефектны по структуре, утратили часть своих поздних репликативных генов и приобрели взамен видоизмененные клеточные гены-V-онкогены, которые и вызывают неопластическую трансформацию при повторной интеграции в клеточный геном. Для полного цикла репликации этим вирусом требуются вирусы-помощники. Клеточные протоонкогены являются прототипами V-онкогенов, консервативными регуляторами клеточной дифференцировки.
Встраивание быстро-трансформирующего реторовируса может либо привести к экспрессии в клетке V-онкогена, либо вирусные промоторы и энхансеры встраиваются рядом с протоонкогенами клетки, вызывая их экспрессию.
Таким образом, встраивание ретровирусов в геном клетки приводит к гиперэкспрессии протоонкогенов, переход их в онкогены с последующей малигнизацией клетки [20, 23, 30, 64].
Что касается механизмов индукции неоплазий химическими канцерогенами с точки зрения современных теорий канцерогенеза – протоонкогенов, онкогенов, антионкогенов, то необходимо остановиться на анализе лишь некоторых работ, посвященных данной проблеме.
Как известно, химические канцерогены, подобно биологическим, способны вызывать развитие мутаций и активацию протоонкогенов [25, 64]. Под влиянием химических канцерогенов возможна онкогенная трансформация в процессе амплификации ДНК. Установлено, что амплификация гена резистентности на фоне воздействия цитостатиков нередко возникает при раке кишечника и является причиной устойчивости неоплазий к химиотерапии. При ряде онкологических заболеваний желудочно-кишечного тракта возникает амплификация онкогенов erbB2, mys, SRS. Индукция развития опухолей нитрозмочевиной связана с амплификацией и активацией N-ras; в опухолях, индуцированных гамма-облучением, активен Ras-H. В ходе химического канцерогенеза отмечено гипометилирование протоонкогена Ras-H, приводящего к развитию генной мутации.
В опухолях, индуцированных химическими канцерогенами, отмечены транскрипции ряда других онкогенов (c-ras и c-mys), связанные с гипометилированием протоонкогена либо его амплификацией. В ходе химического канцерогенеза нарушается зависимость экспрессии c-mys (но не c-ras) от клеточного цикла. Таким образом, многие химические соединения или физические воздействия, а также вирусы могут вызывать мутации ДНК, не летальные для клеток и провоцирующие экспрессию протоонкогенов или депрессию антипротоонкогенов [108]. Последнее приводит к трансформации нормальной клетки в опухолевую.
Эпигенетический механизм канцерогенеза связан с нарушением регуляции клеточного роста, функции клетки и экспрессии генов без повреждения генома. При эпигенетическом канцерогенном эффекте эндогенных или экзогенных канцерогенных факторов возникает инактивация белков-продуктов антипротоонкогенов или активация пострецепторных передатчиков ростовых факторов. Такое воздействие, как правило, не вызывает неоплазии, но усиливает ростовые эффекты, способствует пролиферации мутантного клона и формированию распознаваемой неоплазии. Эффект канцерогенов-мутагенов называют инициирующим, а коканцерогенов – активирующим.
Таким образом, в настоящее время очевидны следующие механизмы активации протоонкогенов:
1) амплификация протоонкогенов, в результате чего резко возрастает их общая активность, что может привести к малигнизации клетки;
2) мутации протоонкогенов, приводящие к их активации, и ингибиция антипротоонкогенов;
3) транслокация протоонкогенов в локус с функционирующим промотором;
4) аддукция промотора рядом с протоонкогеном. В качестве промотора могут выступать ДНК-копии определенных участков онкорнавирусов, а также мобильные генетические структуры, способные перемещаться и встраиваться в различные участки генома.
В геноме человека предполагается наличие около 100 протоонкогенов, выполняющих следующие функции:
1) кодирование ростовых факторов, их рецепторов и пострецепторных передатчиков;
2) кодирование блокаторов запрограммированной гибели клеток, контактного ингибирования пролиферации.
Трансформация протоонкогенов в онкогены приводит к их экспрессии и синтезу онкобелков. При этом онкобелки продуцируются перманентно в увеличенном количестве или в качественно измененном состоянии.
Ниже представлены несколько групп протоонкогенов, антионкогены, и кодируемые ими белки [30, 31, 32].

Обзор
Малигнизация — один из самых загадочных процессов. Что же на самом деле направляет клетку на тернистый путь перерождения?
Автор
Редакторы



Спонсором приза зрительских симпатий выступила компания BioVitrum.
Гончие еще играют во дворе, но дичи не уйти,
как ни мчится она уже сейчас по лесам.
Франц Кафка
Под малигнизацией понимают приобретение здоровыми клетками черт злокачественности, которые мы подробно рассмотрим ниже. Процесс злокачественного изменения можно уподобить дичи из цитаты Кафки, ведь клетка, однажды встав на этот путь, не сможет вернуться и получить свое клеточное здоровье обратно. Важную роль в понимании основ перерождения клеток и их дальнейшего функционирования сыграла медицина, а следом за ней и молекулярная биология. Но начнем с истоков истории рака.
Часть 1. Биография рака
Первые упоминания о раке встречаются в папирусе Эдвина Смита, датируемом 16 веком до нашей эры [1]. Там же отмечается, что данное заболевание не поддается лечению.
Во времена Гиппократа, около 400 года до нашей эры, появилось специальное обозначение рака — karkinos. Разросшаяся опухоль напомнила Гиппократу краба, окутывающего все вокруг клешнями. Современное название онкологии произошло от греческого слова onkos, которое греки использовали для описания опухолей. Однако врачи того времени не различали доброкачественные и злокачественные новообразования, и karkinos Гиппократа не имеет ничего общего с истинным раком.
Гиппократ выдвинул гуморальную теорию, суть которой состояла в том, что каждый недуг является следствием переизбытка одного из четырех гуморов: крови, слизи, желтой желчи и черной желчи.
Первая половина XX века породила еще одну теорию канцерогенеза, недалеко ушедшую от истины. В 1911 году Пейтон Раус, работая в Рокфеллеровском университете в Нью-Йорке, открыл вирус, способный вызывать опухоли у кур. Ученые по всему свету бросились искать вирусы, ответственные за рак именно у человека, однако ничего не могли найти. В 1974 году в Medical World News вирус рака у человека ставили в один ряд с НЛО, снежным человеком и лохнесским чудовищем. Вирус СВ-40 и вирус папилломы человека, вызывающие рак у людей, были открыты в 1960 и 1983 годах соответственно.
В 1970 году генетик Говард Темин, работавший в лаборатории Макардла в Висконсине и изучавший вирус саркомы Рауса (ВСР, или VSR), представил свою работу на Десятом Международном онкологическом конгрессе. Он открыл у ВСР обратную транскрипцию — синтез ДНК по РНК — и положил начало изучениям ретровирусов. Позднее он отказался от вирусной теории канцерогенеза, а в 1979 году ученые Майкл Бишоп и Харолд Вармус открыли первый протоонкоген — src (сарк), содержащийся в ВСР. Это положило начало новому этапу в истории онкологии, люди наконец-то поняли, как запускается процесс канцерогенеза. Но этого бы не произошло без изучения раковой клетки и ее странной физиологии.
Часть 2. Что заставляет клетку измениться?
В этой главе мы разберем причины злокачественного перерождения клетки. Первым толчком к началу этого изменения является мутация в ДНК.
Важными факторами, вызывающими мутации и провоцирующими раковое перерождение, являются ионизирующее излучение, воздействие ультрафиолетовых лучей, влияние цитотоксических веществ, повреждающих ДНК (к ним относятся наркотические вещества и некоторые лекарственные препараты — например, цисплатин, повреждающий структуру двойной спирали) и органические яды.
Но не всякие повреждения ДНК обязательно приведут к появлению раковой клетки, а лишь те, что затронут определенные гены. Наиболее важную роль в канцерогенезе играют три группы генов: протоонкогены, онкогены и гены — супрессоры опухолей.
Протоонкогены
Основные изменения, происходящие с протоонкогенами:

Рисунок 1. Химерный ген BCR-ABL образуется при слиянии участка 9 хромосомы, несущей ген ABL, с участком 22 хромосомы, несущей ген BCR
В некоторых случаях канцерогенез запускается вирусами. Онкогены в геноме вирусов являются ранее захваченными в клетках-хозяевах нормальными генами, которые со временем превратились в злокачественные. Когда такие онкогенные вирусы попадают в клетку, начинается считывание информации с вирусной ДНК или РНК, в цитоплазме накапливаются онкогенные белки и начинается процесс перерождения.
Онкогены
Онкогены — это гены, активность которых стимулирует образование и развитие злокачественной опухоли. Как уже было упомянуто выше, первый вирусный онкоген был открыт в 1979 году.
Биохимические продукты онкогенов
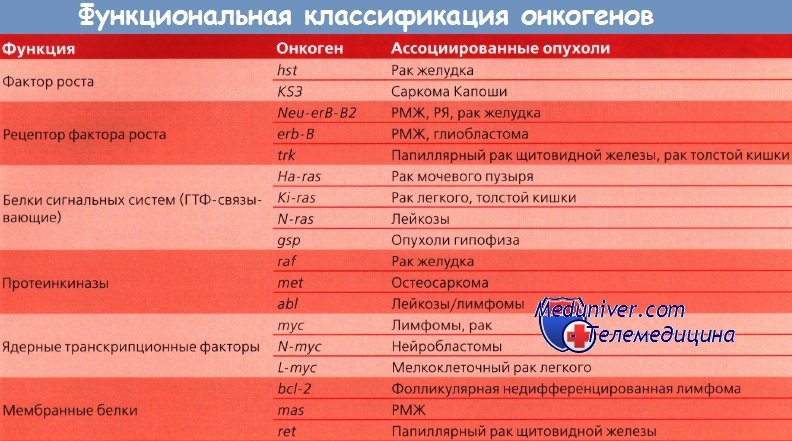
Онкогены кодируют белки с различной структурой и функциями. К основным продуктам деятельности онкогенов относят:
- Факторы роста. Раковые клетки продуцируют белки, способные вызывать пролиферацию и дифференцировку клеток. Наиболее известным фактором роста является HER2, кодируемый геном ERBB2. Мутации и гиперэкспрессия этого гена обнаружены при раке молочной железы и ассоциированы с крайней агрессивностью опухоли. Суперэкспрессия гена приводит к запуску белковых каскадов, ответственных за клеточное деление. Постоянные сигналы к делению вызывают неконтролируемую пролиферативную активность клеток и их злокачественное перерождение.
- ГТФ-связывающие белки. Гуанозинтрифосфат-связывающие белки участвуют во многих клеточных процессах: передача сигналов, транспорт метаболитов внутри клетки и др. Первыми открытыми ГТФ-связывающими белками были белки семейства Ras — продукты онкогена RAS. При постоянном производстве они вызывают злокачественный рост. Наиболее изученный эффектор Ras — это RAF, который запускает белковый каскад MAPK, отвечающий за клеточное деление и пролиферацию [6].
- Мембранные рецепторы. В онкогенезе основную роль играют рецепторы с тирозинкиназной активностью. Они служат для связывания с ростовыми факторами. К ним относится рецептор эпидермального фактора роста, повышенный синтез которого приводит к перерождению клетки.
- Онкогенные протеинкиназы. Протеинкиназы — это группа ферментов, которые модифицируют белки путем фосфорилирования (присоединения остатка фосфорной кислоты). Протеинкиназы регулируют апоптоз, процессы роста и дифференцировки клеток. Нарушения в их работе приводят к сбою в клеточном цикле и, как следствие, к развитию рака. Например, протеинкиназа AKT1, ответственная за ингибирование апоптоза, при перепроизводстве способна вызывать перерождение клеток. Также, она связана с ростом сосудов в опухоли, что помогает раковым клеткам расселяться по организму и давать метастазы.
Все вышеперечисленные продукты онкогенов являются сигналами к запуску неконтролируемого клеточного деления. Внешние факторы больше не играют никакой роли в жизни клетки, потому что пролиферацию запускают внутренние сигнальные белки.
Гены и белки — супрессоры
В здоровой клетке существуют защитные механизмы, следящие за процессами и регулирующие клеточный цикл. К таким механизмам относят деятельность белков — супрессоров опухолей: p21, p53, pRb, PTEN и др.
Белок p53 — наиболее изученный белок-супрессор. Он является продуктом гена TP53, мутации которого обнаруживаются в клетках многих опухолей [7]. p53 синтезируется во всех клетках организма, но активируется только при повреждениях ДНК. Этот белок способен остановить клеточный цикл и не допускать дальнейшее деление клетки, пока не произойдет репарация ДНК. При сильных повреждениях он также может запускать процесс апоптоза.
Одной из главных функций p53 является сохранение генетической идентичности всех клеток организма. При неправильной работе этого белка клетка получает возможность делиться даже при поврежденной ДНК, что увеличивает вероятность мутаций и накопления дефектных онкогенов. Важную роль в подавлении p53 играет белок MDM2, который в норме регулирует активность p53. Однако при повышенном синтезе он связывается с p53 и ингибирует его противоопухолевое действие.
Эпигенетические факторы рака
Важными факторами канцерогенеза являются эпигенетические события. Эпигенетика изучает процессы, затрагивающие активность генов, но не изменяющие структуру ДНК. К ним относится изменение метилирования ДНК.
Метилирование — это присоединение метильной группы к нуклеотидам в особых, строго определенных участках генома, называемых CpG-островками. Такое изменение не влияет на структуру молекулы, однако может влиять на экспрессию отдельных генов. В частности, если в участке ДНК много метильных групп, то транскрипция этого участка прекращается.
Особенно активно метилирование проходит в эмбриональный период жизни, а у взрослого человека метилировано около 2% генома. В норме баланс между метилированием и деметилированием строго регулируется и соблюдается, однако в старости начинают преобладать процессы метилирования, что может в итоге привести к канцерогенезу. В процессе онкогенеза происходит гиперметилирование CpG-островков, что приводит к общей геномной нестабильности и накоплению еще большего количества мутаций. В большинстве случаев метилированные участки являются промоторами и влияют на активацию или, наоборот, инактивацию генов, что с виду похоже на действие точечных мутаций.
Однако хотя нарушения в эпигенетической регуляции сопровождают развитие злокачественного перерождения, они, как правило, не являются его первопричиной, а лишь одним из сопутствующих факторов.

Рисунок 2. Раковый геном. В процессе жизнедеятельности раковая клетка накапливает огромное количество мутаций и нередко характиризуется полиплоидностью.
Часть 3. Физиологические последствия малигнизации
Основное последствие малигнизации — клеточное бессмертие. Оно может поддерживаться несколькими способами: активацией фермента теломеразы, блокировкой регуляторов митохондриального пути апоптоза и в некоторых случаях активацией механизма ALT (alternative lengthening of telomeres, альтернативного удлинения теломер [14]).
Впервые клеточное бессмертие раковых клеток было продемонстрировано в 1951 году на клеточной линии HeLa, взятой у Генриетты Лакс, вскоре скончавшейся от рака шейки матки (рис. 3) [9].

Как правило, малигнизация сопровождается активацией фермента теломеразы. На концах хромосом находятся короткие повторяющиеся участки ДНК, названные теломерами [10]. После каждого деления теломеры укорачиваются, что в итоге приводит к их полному исчезновению и невозможности продолжать деление. Количество возможных делений для клетки названо пределом Хейфлика. Действие теломеразы заключается в восстановлении теломер и превращении клетки в фактически бессмертную, позволяя ей делиться бесконечно долго. Существуют нормальные клетки, в которых также экспрессируется теломераза. Это клетки, которым надо часто делиться: половые, стволовые и клетки эпителия кишечника. Однако теломераза активна в подавляющем большинстве раковых клеток, что играет важную роль в их жизненном цикле.
С другой стороны, в некоторых злокачественных клетках, наравне с активной теломеразой, существует так называемое альтернативное удлинение теломер, или сокращенно ALT [11]. При ALT происходит гомологичная рекомбинация концевых участков хромосом (рис. 4). В норме рекомбинация происходит в процессе мейоза, однако раковые клетки научились достраивать теломеры, используя теломеры другой хромосомы как матрицу [12].

Важно отметить, что раковое бессмертие контролируется не только теломерами, но и ингибированием путей апоптоза, главным из которых является митохондриальный путь. В норме, из митохондрий в цитоплазму выходят митохондриальные белки и образуют апоптотический комплекс — апоптосому, которая и запускает апоптоз. При неправильной работе регуляторных белков, а к ним относятся белки семейства BCL-2, нарушается выход апоптотических белков, что приводит к сбою в процессе апоптоза. В раковых клетках обнаружены нарушения в работе белков BAX и BAK, а также экспрессия ингибиторов клеточной смерти.
Часть 4. Заключение
Читайте также: