
Поражения легких при заболеваниях соединительной ткани реферат
Обновлено: 04.07.2024
Не существует адекватного определения, которое охватывало бы группу болезней, из которых каждая имеет собственные, довольно характерные клинико-патологические признаки, причем при всех болезнях могут поражаться многие органы, включая легкие. Коллагеновый некроз и васкулит встречаются часто, но не всегда.
По отдельности эти заболевания представляют собой весьма сложные проблемы в пульмонологии, так как патологоанатомические, клинические и иммунологические признаки многих из них образуют сложные переплетения. Вследствие этих трудностей клиницисты и ученые высказывают различные предположения о патогенезе вообще и иммунопатогенезе в частности, часто в отсутствие фактических данных, относящихся к сыворотке, клеткам или тканям отдельных больных.
Действительно, в некоторых условиях прямое наблюдение отдельных больных опровергает предложенные иммунологические гипотезы. Ввиду больших пробелов в наших познаниях в настоящее время, быть может, было бы целесообразно по возможности подробно изложить сделанные наблюдения и выждать дальнейшей информации, прежде чем переходить к каким-либо обобщениям о патогенезе.
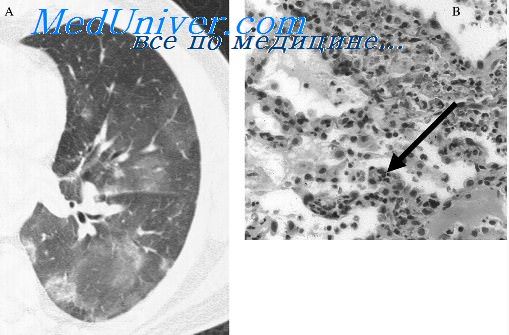
Легочную патологию можно разделить на пять основных типов. Во всех этих случаях может наблюдаться вовлечение легких при генерализованном заболевании, когда страдают и другие системы, либо легкие могут быть единственным пораженным органом. В этих рамках можно объединить большое количество общеизвестных клиникопатологических синдромов.
При многих из них патологические поражения могут иметься в ряде органов, но только в некоторых органах обнаруживаются признаки васкулита. Хорошо известными примерами служат системная красная волчанка, ревматоидный артрит пли узелковый полиартериит, когда в некоторых тканях можно видеть очаги васкулита, а в других только хронический воспалительный экссудат.
В некоторых случаях можно видеть аутоантитела, особенно неорганоспецифические (антиядерные, ревматоидный фактор и антитела против базальных мембран), однако в других случаях их нет. Они особенно часто встречаются в группе больных с неэозипофильными инфильтратами и клинически выраженным васкулитом или без него.
Однако очень трудно установить, играют ли они какую-то роль в патогенезе и какую именно. Предложенная классификация свидетельствует также о том, что у больных с характерными внелегочными синдромами, например системной красной волчанкой или ревматоидным артритом, в легких может быть только один тип из ряда различных патологических изменений. Например, ревматоидный артрит может сочетаться с изолированным некробиотическим узелком в легких у одного больного и широко распространенным фиброзирующим альвеолитом у другого больного.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Что такое поражение легких при диффузных болезнях соединительной ткани -
Что провоцирует / Причины Поражений легких при диффузных болезнях соединительной ткани:
Поиски специфического инфекционного агента оказались безрезультатными, за исключением ревматизма, при котором можно считать доказанной этиологическую роль гемолитического стрептококка группы А (в особенности при острых формах, протекающих с поражением суставов).
Попытки объяснить причину возникновения заболеваний различными нейрогенными и (или) эндокринными нарушениями также не получили достаточного фактического подтверждения.
Господствующей в последние годы является вирусная теория этиологии, которая обсуждается практически при всех диффузных болезнях соединительной ткани. Предполагается, что воздействие на организм различных патогенных факторов (аллергических, инфекционных, токсических и др.) приводит к активизации латентно персистирующего вируса в организме, что, в свою очередь, приводит к различной степени выраженности-иммунных и аутоиммунных сдвигов, нарушениям коллагенового обмена, микроциркуляторным нарушениям.
Патогенез (что происходит?) во время Поражений легких при диффузных болезнях соединительной ткани:
Обнаружение в пораженных тканях вирусоподобных включений ирн системной склеродермии, дсрматомиозите, синдроме Шегрена, а также своеобразных тубулоретикулярных вирусоподобных включений в эндотелии капилляров пораженных тканей при системной красной волчанке, австралийского антигена у части больных узелковым периартериитом говорит в пользу вирусной теории этиологии диффузных болезней соединительной ткани. Отмечено, что иммунологическая перестройка, происходящая в организме при узелковом периартериите, может вызываться В-вирусом гепатита. Развитие узелкового периартериита в этом случае связывается с австралийским антигеном и антителами к нему. Провоцирующую роль могут играть различные физические факторы: ионизирующая радиация, инсоляция, переохлаждение и др. Выявление в стенках пораженных сосудов флюоресцирующих антител, иммунных комплексов и углобулинов свидетельствует об иммунологических нарушениях при этом заболевании. В качестве подтверждающих вирусную теорию фактов следует отметить выявление у больных коллагено-зами маркеров персистирующей вирусной инфекции: лимфоци-тотоксических антител, антител к двусииральной РНК, повышение титра противовирусных антител и др.
Очевидно, вирусная теория не исключает (а скорее предполагает) участие в патогенезе иммунологических, токсических и других механизмов. Исследования последних лет показали, что нарушения в системе иммунитета -один из наиболее постоянных признаков диффузных болезней соединительной ткани. Так, при системной красной волчанке важное значение в патогенезе отводится выявленным аномалиям в системе клеточного и гуморального иммунитета. Предполагается, что угнетение выработки Т-лимфоцитов (в частности, Т-супрессоров) вирусами и лимфоцитотоксичсскими антителами приводит к повышенной продукции иммуноглобулинов В-клетками. Отмечена зависимость между активностью патологического процесса и уровнем В-лимфоцитов. Гиперпродукция аутоантител ведет к образованию циркулирующих иммунных комплексов, осаждению их на базальиы-х мембранах сосудов, в первую очередь капилляров. Повреждающее действие иммунных комплексов проявляется развитием васкулита и микроциркуляторными нарушениями в почках, коже, легких и других органах. Деструкция соединительной ткани приводит к поступлению в кровоток новых антигенов, что, в свою очередь, стимулирует образование новых иммунных комплексов.
Выраженные иммунологические сдвиги при диффузных болезнях соединительной ткани подтверждаются частым выявлением в крови у этих больных высоких титров ревматоидного фактора, противоядерных антител (в особенности при ревматоидиом артрите и синдроме Шегрена), гипергаммаглобулине--мии, снижения уровня комплемента, повышения уровня сывороточного IgG и др. Характерные для системной красной волчанки и узелкового периартериита клинические и серологические признаки могут возникать при длительном приеме больными некоторых препаратов: пенициллина, сульфаниламидов, при повторном введении сывороток, вакцин, длительном приеме контрацептивных средств.
Важное значение в патогенезе диффузных болезней соединительной ткани отводится в последние годы семейно-генетическому предрасположению. Так, ревматоидный артрит встречается в 2-10 раз чаще в семьях, где есть другой больной ревматоидным артритом. Описаны семейные формы системной склеродермии, системной красной волчанки, ревматизма и других коллагенозов. При системной склеродермии важная роль в развитии фиброзпо-склеротических изменений отводится продукции аномального коллагена (увеличение биосинтеза коллагена III типа) вследствие генетических дефектов. Это позволяет относить системную склеродермию к классическим (истинным) коллагенозам [Насонова В. А., 1978, и др.].
Преимущественный тип патоморфологических изменений в легких зависит от вида коллагеноза и формы течения болезни (острая,подострая, хроническая). При остром течении более характерно поражение легких по типу васкулита, при хроническом - по типу интерстициального пневмонита. Легочный васкулит характеризуется деструктив и опролифератным процессом в стенках ветвей легочной и бронхиальной артерий с фибриноидным некрозом, тромбозом и развитием аневризм пораженных сосудов (некротизирующий ангиит), кровоизлияниями в паренхиму легких. Образование полостей один из характерных признаков легочного васкулита. Поражение легких по типу васкулита более характерно для узелкового периартериита, острых форм системной красной волчанки, дерматомио-зита, встречается при ревматоидном артрите, синдроме Шегрена и менее характерно для системной склеродермии.
Характерными патологоанатомическими особенностями отдельных форм коллагеновых болезней являются поражения плевры с выпотом в плевральную полость (как проявление синдрома полисерозита) при системной красной волчанке, сухой или выпотной плеврит с наличием ревматоидных гранулем на плевре и в паренхиме легких при ревматоидном артрите н ревматизме, поражение межреберных мышц и диафрагмы при дермато-миозите, плеврофиброз и разрывы субплевральных кист при системной склеродермии. Наиболее характерным патоморфологи-ческим признаком синдрома Шегрена является инфильтрация лимфоцитами и плазматическими клетками экзокринных желез в первую очередь слезных и слюнных, а также слизистых желез трахеи и бронхов. Функционирующая паренхима желез постепенно замещается фиброзной тканью. Атрофический процесс в слизистой оболочке трахеи и бронхов ведет к нарушению мукоцилиарного клиренса, снижению уровня секреторного IgA, что предрасполагает к присоединению вторичной инфекции (хронический бронхит, трахеит, пневмония). Присоединение вторичной инфекции на фоне основного заболевания встречается и при других коллагенозах. Для дерматомиозита более характерна аспирационная пневмония.
Симптомы Поражений легких при диффузных болезнях соединительной ткани:
Выраженность и тяжесть респираторных проявлений зависят от степени агрессивности основного процесса. При поражении легких по типу интерстициального пневмонита начальные признаки болезни могут быть стертыми или проявляться одышкой, кашлем (как правило, сухим или со скудной слизистой мокротой), болями в грудной клетке. Одышка носит прогрессирующий характер.
Кровохарканье, легочные кровотечения указывают на явления васкулита в легочной ткани. Плевральные экссудаты не только наиболее частая форма плевролегочных проявлений ревматоидного артрита, ревматизма, системной красной волчанки, но нередко и первый признак заболевания. В 12--25 % случаев узелкового периартериита выявляется бронхоспастический синдром. При синдроме Шегрена нередко больные жалуются на охриплость голоса (следствие сухости слизистых оболочек, голосовых связок). Наряду с этим выявляются характерные признаки этой болезни: керато-конъюнктивит, сухость слизистых оболочек рта, носа, глотки, половых органов, нарушение секреторной функции желудочно-кишечного тракта, синдром Рейно.
Перкуторный тон над нижними отделами легких при наличии интерстициального пневмонита укорочен. Могут выслушиваться крепитирующие хрипы, ослабленное везикулярное дыхание. Жесткое дыхание, рассеянные сухие хрипы более характерны для синдрома Шегрена, сухие свистящие хрнпы выслушиваются при наличии бронхоспастнческого синдрома узелкового периартериита.
Диагностика Поражений легких при диффузных болезнях соединительной ткани:
На рентгенограммах интерстициальный пневмонит определяется в виде усиления легочного рисунка преимущественно за счет интерстициального компонента. При обострении болезни может усиливаться сосудистый компонент легочного рисунка. Высокое стояние куполов диафрагмы, дисковидные ателектазы, прогрессирующий фиброз легких - наиболее характерные рентгенологические признаки интерстициального пневмонита при диффузных болезнях соединительной ткани. Дисковидные ателектазы наиболее часто встречаются при дерматомиозите и системной склеродермии. Иногда определяются кальцификаты в плевре.
Следует отметить, что .перечисленные рентгенологические признаки не патогномоничны для интерстициального пневмонита при коллагеновых болезнях. Они могут наблюдаться и при идио-патическом фиброзирующем альвеолите, экзогенных аллергических фиброзирующих альвеолитах, токсических фиброзирую-щих альвеолитах и других, более редких заболеваниях.
Для легочного васкулита более характерны затенения по типу пйевмонии или в виде мелких очагов. При поражении более крупных сосудов развивается клинико-рентге-нологическая картина инфаркта легкого, нередко-с образованием полостей [Розешптраух Л. С. и др., 1978].
Нарушения вентиляционной способности легких, как правило, предшествуют рентгенологическим изменениям. Рестрик-тивный тип нарушения вентиляции, снижение диффузионной способности легких, снижение статической растяжимости легких - характерные, но не патогномоничные признаки интерстициального пневмонита. Обструктивные нарушения вентиляционной способности легких являются ведущими при бронхоспастическом синдроме узелкового периартериита.
Диагностика и дифференциальная диагностика поражения легких при наличии четкой клинико-лабораторной картины кол-лагеноза не вызывают особых затруднений. Однако следует учитывать, что поражение легких иногда может быть первым проявлением коллагеноза; нередко клинические проявления коллагеноза (в особенности на ранних этапах заболевания) могут не укладываться в рамках конкретной нозологической формы.
При проведении дифференциальной диагностики необходимо в первую очередь исключить первичный опухолевый процесс (бронхиоло-альвеолярный рак) или метастазы опухоли другой локализации. Отмечено, что заболеваемость раком у больных дерматомиозитом в 8 раз выше, чем у остального населения. Это вызвало предположение, что дерматомиозит нередко является синдромом основного опухолевого процесса. Дерматомиозит следует отличать также от прогрессирующей мышечной дистрофии, тиреотоксической миопатии.
Дифференциальная диагностика поражения легких при кол-лагенозах проводится с туберкулезом легких, иднопатическпм фпброзпрующим альвеолитом, экзогенными аллергическими фнброзирующимн альвеолитами, токсическим фиброзирующим альвеолитом, инфарктпневмонией, а при наличии бронхоспаз-ма - с бронхиальной астмой.
При подозрении на узелковый периартериит дифференциальную диагностику следует проводить также с аллергическим гранулематозным ангиитом, впервые описанным в 1951 г. Churg и Strauss. Клинически синдром Чердж - Строе характеризуется тяжелыми приступами удушья, гиперэозипофилией крови, высокой лихорадкой, прогрессирующей сердечной недостаточностью, болями в брюшной полости, кожными и неврологическими поражениями. При гистологическом исследовании наряду с изменениями, характерными для узелкового пернартерии-та, выявляется поражение артериол и вепул с гпгантоклеточпы-мн внесосудистымн гранулемами, что является патоморфологи-ческой особенностью этого синдрома.
Диагностика и дифференциальная диагностика синдрома Шегрена основывается на выявлении характерных для этого заболевания клинических признаков (кератоконъюнктивит, ксеростомня, полиартрит, синдром Рейно), выраженных аутоиммунных сдвигов, характерных патоморфологических изменений при исследовании материала биопсий слюнных желез, слизистых оболочек.
Течение. Для диффузных болезней соединительной ткани характерно прогрессирующее течение. Присоединение вторичной инфекции - одно из характерных осложнений при прогрессировании патологического процесса в легких. Тяжелые осложнения (аспирационная пневмония, асфиксия, гипостатическая пневмония) нередко наблюдаются при дерматомиознте и системной склеродермии. Легочный васкулит может осложняться образованием полостей, легочными кровотечениями, формированием абсцесса легких, эмпиемы плевры. Синдром Шегрена нередко осложняется рецидивирующим плевритом, перикардитом, сахарным диабетом, хроническим панкреатитом, очаговым гломерулонефритом, нефрокальцинозом и др. Одним из напбиолее характерных осложнений синдрома Шегрена является образование лимфом и псевдолпмфом.
Лечение Поражений легких при диффузных болезнях соединительной ткани:
Лечение каждой из форм коллагеновых болезней имеет свои особенности. Общим для этой группы заболеваний является возможность получения положительного эффекта при назначении кортикостероидных препаратов, а при их недостаточной эффективности или плохой переносимости - в сочетании с иммуносупрессорами (азатиоприн, циклофосфамид, метотрексат, б-меркаптопурин) или купренилом. Препаратами выбора все же остаются кортикостероиды. После получения терапевтического эффекта первоначальную дозу гормонов (1- 1,5 мг на 1 кг массы тела) снижают до поддерживающей (5- 15 мг в сутки). Длительность лечения и дозы зависят от нозологической формы, получаемого эффекта. Комплексная терапия включает также поливитамины, противовоспалительные средства, анаболические гормоны, лечебную физкультуру и физиотерапевтические процедуры. Антибиотики и сульфаниламиды показаны только при наличии вторичной инфекции. Больным с синдромом Шегрена рекомендуется назначение диметилсульфо-ксида в комплексе с гепарином, гидрокортизоном, аскорбиновой кислотой.
Прогноз. Своевременное лечение кортикостероидами в комплексе с другими препаратами позволяет продлить жизнь этой категории больных. Присоединение вторичной инфекции (абсцесс легких, эмпиема плевры, легочные кровотечения) ускоряет летальный исход.
К каким докторам следует обращаться если у Вас поражение легких при диффузных болезнях соединительной ткани:
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Поражений легких при диффузных болезнях соединительной ткани, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору .
Частота и варианты поражения органов дыхания при системных заболеваниях соединительной ткани (СЗСТ) существенно различаются, оказывая значительное влияние на их клинические особенности и прогноз. Легочные поражения встречаются у 20–95 % больных СЗСТ, при этом состояния могут различаться – от бессимптомных до жизнеугрожающих, вплоть до летального исхода. Нередко осложнения, обусловленные интерстициальным заболеванием легких (ИЗЛ), на фоне СЗСТ становятся ведущими в определении прогноза у больных. Интерстициальные пневмонии являются самым частым вариантом поражения респираторной системы при СЗСТ. Среди всех ИЗЛ на долю СЗСТ-ассоциированных приходится от 15 до 25 % случаев. Чаще поражение органов дыхания формируется в период развернутой клинической картины СЗСТ. Однако в некоторых случаях СЗСТ могут дебютировать как ИЗЛ, предшествуя системным проявлениям болезни, что существенно затрудняет раннюю нозологическую диагностику. В таких случаях физикальное обследование пульмонологом должно включать поиск возможных экстрапульмональных проявлений СЗСТ. Кроме того, неотъемлемой частью диагностического алгоритма является специфическое лабораторное обследование – определение уровня аутоантител, которое при сопоставлении с клинической картиной обеспечивает высокую степень вероятности диагноза СЗСТ.
Ключевые слова
Об авторах
Трофименко Ирина Николаевна – доктор медицинских наук, доцент кафедры клинической аллергологии и пульмонологии.
664079, Иркутск, мкрн Юбилейный, 100.
тел.: (9148) 77-80-96
Черняк Борис Анатольевич – доктор медицинских наук, профессор, заведующий кафедрой клинической аллергологии и пульмонологии.
664079, Иркутск, мкрн Юбилейный, 100.
Список литературы
1. Jokerst C., Purdy H., Bhalla S. An overview of collagen vascular disease-associated interstitial lung disease. Semin. Roentgenol. 2015; 50 (1): 31–39. DOI: 10.1053/j.ro.2014.04.006.
2. Cottin V. Idiopathic interstitial pneumonias with connective tissue diseases features: a review. Respirology. 2016; 21 (2): 245–258. DOI: 10.1111/resp.12588.
3. Das S., Padhan P. An Overview of the extraarticular involvement in rheumatoid arthritis and its management. J. Pharmacol. Pharmacother. 2017; 8 (3): 81–86. DOI: 10.4103/jpp.JPP_194_16.
4. De Lauretis A., Veeraraghavan S., Renzoni E. Review series: Aspects of interstitial lung disease: connective tissue disease-associated interstitial lung disease: how does it differ from IPF? How should the clinical approach differ? Chron. Respir. Dis. 2011; 8 (1): 53–82. DOI: 10.1177/1479972310393758.
5. Spagnolo P., Cordier J.F., Cottin V. Connective tissue diseases, multimorbidity and the ageing lung. Eur. Respir. J. 2016; 47 (5): 1535–1558. DOI: 10.1183/13993003.00829-2015.
6. Vij R., Strek M.E. Diagnosis and treatment of connective tissue disease-associated interstitial lung disease. Chest. 2013; 143 (3): 814–824. DOI: 10.1378/chest.12-0741.
7. Jee A.S., Adelstein S., Bleasel J. et al. Role of autoantibodies in the diagnosis of connective-tissue disease ILD (CTD-ILD) and interstitial pneumonia with autoimmune features (IPAF). J. Clin. Med. 2017; 6 (5): pii: E51. DOI: 10.3390/jcm6050051.
8. Koo S.M., Uh S.T. Treatment of connective tissue disease-associated interstitial lung disease: the pulmonologist's point of view. Korean J. Intern. Med. 2017; 32 (4): 600–610. DOI: 10.3904/kjim.2016.212.
9. Wallis A., Spinks K. The diagnosis and management of interstitial lung diseases. Br. Med. J. 2015; 350: h2072. DOI: 10.1136/bmj.h2072.
11. Valeyre D., Duchemann B., Nunes H. et al. Interstitial lung diseases. Respiratory Epidemioloogy. ERS Monogr., chapter 6. 2014; 65: XIV–XVII.
12. Mathai S.C., Danoff S.K. Management of interstitial lung disease associated with connective tissue disease. Br. Med. J. 2016; 352: h6819. DOI: 10.1136/bmj.h6819.
13. Antoniou K.M., Margaritopoulos G., Economidou F. et al. Pivotal clinical dilemmas in collagen vascular diseases associated with interstitial lung involvement. Eur. Respir. J. 2009; 33 (4): 882–896. DOI: 10.1183/09031936.00152607.
14. Fischer A., du Bois R. Interstitial lung disease in connective tissue disorders. Lancet. 2012; 380 (9842): 689–698. DOI: 10.1016/S0140-6736(12)61079-4.
15. Tzelepis G.E., Toya S.P., Moutsopoulos H.M. Occult connective tissue diseases mimicking idiopathic interstitial pneumonias. Eur. Respir. J. 2008; 31 (1): 11–20. DOI: 10.1183/09031936.00060107.
16. Mittoo S., Gelber A.C., Christopher-Stine L. et al. Ascertainment of collagen vascular disease in patients presenting with interstitial lung disease. Respir. Med. 2009; 103 (8): 1152–1158. DOI: 10.1016/j.rmed.2009.02.009.
17. Duchemann B., Annesi-Maesano I., Jacobe de Naurois C. et al. Prevalence and incidence of interstitial lung diseases in a multi-ethnic county of Greater Paris. Eur. Respir. J. 2017; 50 (2): pii: 1602419. DOI: 10.1183/13993003.02419-2016.
18. Strange C., Highland K.B. Interstitial lung disease in the patient who has connective tissue disease. Clin. Chest Med. 2004; 25 (3): 549–559. DOI: 10.1016/j.ccm.2004.05.009.
20. Kim E.J., Collard H.R., King T.E. Jr. Rheumatoid arthritis-associated interstitial lung disease: the relevance of histopathologic and radiographic pattern. Chest. 2009; 136 (5): 1397–1405. DOI: 10.1378/chest.09-0444.
21. Park J.H., Kim D.S., Park I.N. et al. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am. J. Respir. Crit. Care Med. 2007; 175 (7): 705–711. DOI: 10.1164/rccm.200607-912OC.
22. Romagnoli M., Nannini C., Piciucchi S. et al. Idiopathic nonspecific interstitial pneumonia: an interstitial lung disease associated with autoimmune disorders? Eur. Respir. J. 2011; 38 (2): 384–391. DOI: 10.1183/09031936.00094910.
23. Karam M.B., Peivareh H., Mosadegh L. Thoracic imaging findings of collagen vascular diseases: a CT study. Tanaffos. 2014; 13 (1): 43–47.
24. Sato S., Hoshino K., Satoh T. et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: Association with rapidly progressive interstitial lung disease. Arthritis Rheum. 2009; 60 (7): 2193–2200. DOI: 10.1002/art.24621.
25. Gayraud M. Raynaud's phenomenon. Joint Bone Spine. 2007; 74 (1): e1–8. DOI: 10.1016/j.jbspin.2006.07.002.
26. Ungprasert P., Crowson C.S., Chowdhary V.R. et al. Epidemiology of mixed connective tissue disease, 1985–2014: a population-based study. Arthritis Care Res. (Hoboken). 2016; 68 (12): 1843–1848. DOI: 10.1002/acr.22872.
32. Raghu G., Collard H.R., Egan J.J. et al. An off icial ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011; 183 (6): 788–824. DOI: 10.1164/rccm.2009-040GL.
33. Solomon D.H., Kavanaugh A.J., Schur P.H. Evidence-based guidelines for the use of immunologic tests: antinuclear antibody testing. Arthritis Rheum. 2002; 47 (4): 434–444. DOI: 10.1002/art.10561.
34. Giles J.T., Danoff S.K., Sokolove J. et al. Association of fine specificity and repertoire expansion of anticitrullinated peptide antibodies with rheumatoid arthritis associated interstitial lung disease. Ann. Rheum. Dis. 2014; 73 (8): 1487–1494. DOI: 10.1136/annrheumdis-2012-203160.
35. Yin Y., Liang D., Zhao L. et al. Anti-cyclic citrullinated peptide antibody is associated with interstitial lung disease in patients with rheumatoid arthritis. PLoS One. 2014; 9 (4): e92449. DOI: 10.1371/journal.pone.0092449.
36. Fischer A., Antoniou K.M., Brown K.K. et al. An official European Respiratory Society/American Thoracic Society research statement: Interstitial pneumonia with autoimmune features. Eur. Respir. J. 2015; 46 (4): 976–987. DOI: 10.1183/13993003.00150-2015.
37. Tashkin D.P., Elashoff R., Clements P.J. et al. Cyclophosphamide versus placebo in scleroderma lung disease. N. Engl. J. Med. 2006; 354 (25): 2655–2666. DOI: 10.1056/NEJMoa055120.
38. Tashkin D.P., Roth M.D., Clements P.J. et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS-II), a randomised controlled, double-blind, parallel group trial. Lancet Respir. Med. 2016; 4 (9): 708–719. DOI: 10.1016/S2213-2600(16)30152-7.
39. Saunders P., Tsipouri V., Keir G.J. et al. Rituximab versus cyclophosphamide for the treatment of connective tissue disease-associated interstitial lung disease (RECITAL): study protocol for a randomised controlled trial. Trials. 2017; 18 (1): 275. DOI: 10.1186/s13063-017-2016-2.
40. Huang H., Feng R.E., Li S. et al. A case report: The efficacy of pirfenidone in a Chinese patient with progressive systemic sclerosis-associated interstitial lung disease: A CARE-compliant article. Medicine (Baltimore). 2016; 95 (27): e4113. DOI: 10.1097/MD.0000000000004113.
41. Miura Y., Saito T., Fujita K. et al. Clinical experience with pirfenidone in five patients with scleroderma-related interstitial lung disease. Sarcoidosis Vasc. Diffuse Lung Dis. 2014; 31 (3): 235–238.
Рассмотрены вопросы поражения легких при системной красной волчанке, ревматоидном артрите, системной склеродермии, полимиозите/дерматомиозите, синдроме Шегрена, смешанном заболевании соединительной ткани, гранулематозе Вегенера, синдроме Черджа–Стросса, с
Aspects of lungs lesion with exanthematous lupus erythematosus, rheumatoid arthritis, Addison’s keloid, polymyositis/dermatomyositis, Sj?gren’s sicca syndrome, mixed connective tissue disease, necrotizing respiratory granulomatosis, Churg–Strauss syndrome, Goodpasture’s syndrome and Bechterew’s disease have been analysed.
В основе поражения легких (ПЛ) при системных аутоиммунных заболеваниях (САЗ) лежат расстройства механизмов иммунорегуляции и гиперреактивность организма. Макрофаги и лимфоциты являются ключевыми клетками, участвующими в инициации и сохранении иммунного ответа в легких. Альвеолярные макрофаги — резиденты легочной ткани — поглощают чужеродные агенты, проникшие через слизистые поверхности легких и бронхов. Кроме того, эти клетки служат в качестве антигенпредставляющих клеток для Т-лимфоцитов. Относительно небольшое число лимфоцитов присутствуют в нормальной паренхиме легких. Однако после стимуляции соответствующим антигеном окружающей лимфоидной ткани лимфоциты мигрируют в легкие и принимают участие в воспалительной реакции.
Проведение специальных исследований, включавших компьютерную томографию высокого разрешения (КТВР) и биопсию легкого, выявило, что при САЗ встречаются различные гистологические формы поражения интерстиция легких (табл.).
Системная красная волчанка
Системная красная волчанка (СКВ) — системное аутоиммунное заболевание неизвестной этиологии, характеризующееся гиперпродукцией органоспецифических аутоантител к различным компонентам клеточного ядра с развитием иммуновоспалительного повреждения тканей и внутренних органов.
Ревматоидный артрит
Ревматоидный артрит (РА) — системное аутоиммунное заболевание соединительной ткани с преимущественным поражением суставов по типу хронического прогрессирующего эрозивно-деструктивного полиартрита и внесуставными проявлениями.
ПЛ при РА было впервые описано в 1948 г., когда Эллман и Болл установили диффузный фиброз легких у трех пациентов. Легочные проявления при РА включают поражение плевры, образование ревматоидных узелков в легких, ИПЛ, легочный васкулит, альвеолярные кровоизлияния, бронхообструктивные изменения. Почти у половины больных РА при вскрытии выявляется плеврит. Экссудативный плеврит может быть односторонним или двусторонним и сохраняться в течение многих месяцев. Наличие плеврального выпота вызывает нарушение легочной функции. Как правило, плевральная жидкость бывает экссудативной с низким содержанием глюкозы и низким уровнем комплемента. Чаще у пациентов с РА встречается сухой плеврит и является находкой при рентгенологическом исследовании.
Ревматоидные узелки (РУ), как единичные, так и множественные, могут быть обнаружены в легочной паренхиме больных РА. РУ легких у пациентов с РА были впервые описаны Каплан в 1953 г. РУ могут появиться до, во время или после начала РА. ИПЛ характеризуется хроническим воспалением стенок альвеол и скоплением больших мононуклеарных клеток в альвеолах. Довольно часто у больных РА встречается сочетание ИПЛ и подкожных ревматоидных узелков. Прогноз для пациентов с РА с ИПЛ неблагоприятен. Легочный васкулит является одним из редких легочных проявлений РА. Альвеолярные кровоизлияния, не часто встречающиеся у больных РА, приводят к кровохарканью и анемии. У пациентов с РА отмечается высокая частота поражения бронхов. ПЛ может быть результатом токсического воздействия препаратов, используемых в терапии РА. Для того чтобы обеспечить оптимальный эффект от лечения, врач всегда должен учитывать возможную легочную патологию при оценке состояния пациентов с РА [1–3].
Системная склеродермия
Системная склеродермия (ССД) — диффузное заболевание соединительной ткани с прогрессирующим фиброзом, распространенными вазоспастическими нарушениями и характерными изменениями кожи, опорно-двигательного аппарата и внутренних органов. Среди висцеральных проявлений ССД ПЛ занимает важное место.
Плеврит при ССД наблюдается реже, чем при РА и СКВ. В то же время у больных ССД значительно выше частота ИПЛ, при этом морфологическое исследование показывает выраженный фиброз интерстициальной ткани и утолщение межальвеолярных перегородок. Исследование функции внешнего дыхания (ФВД) демонстрирует снижение диффузионной способности легких (ДСЛ) даже при отсутствии каких-либо клинических симптомов и рентгенологических изменений.
У пациентов с ССД выявляется рестриктивный тип нарушения вентиляции легких. У большинства больных ССД с ЛФ наблюдается гистологическая картина обычной интерстициальной пневмонии (ОИП), однако во многих случаях встречается гистопатологический тип неспецифической интерстициальной пневмонии (НИП).
Развитие ЛФ инициируется микрососудистыми изменениями, которые приводят к повреждению эндотелиальных клеток и поражению альвеолярного эпителия. Это приводит к активации каскада свертывания (рис.).
Экспрессия аутоантител является предиктором поражения внутренних органов, в частности ПЛ. Наличие антител к Scl-70 (антисклеродермальные антитела с молекулярной массой 70 кДа, антитела к топоизомеразе I) в значительной степени является фактором риска развития ИПЛ, в то время как антицентромерные антитела (АЦА) говорят о низкой вероятности развития рентгенологических признаков ЛФ.
У пациентов с ССД встречается легочная гипертензия (ЛГ). ЛГ может быть изолированной, возникшей вследствие поражения сосудов, или вторичной — при поражении паренхимы легких или левых отделов сердца. ЛГ развивается у 5–7% больных, чаще на поздних стадиях лимитированной формы ССД. Предиктор ЛГ — изолированное уменьшение ДСЛ. Более редко встречаются аспирационная пневмония, причиной которой является дисфункция пищевода, а также спонтанный пневмоторакс, лекарственный пневмонит [1–3, 6–8].
Полимиозит/дерматомиозит
Полимиозит/дерматомиозит (ПМ/ДМ) — группа хронических диффузных заболеваний поперечнополосатой мускулатуры, основным проявлением которых выступает мышечная слабость.
Около 40% пациентов с ПМ/ДМ имеют патологию легочной системы. Одной из причин этого является поражение мышц. Поражение межреберных мышц и высокое стояние диафрагмы приводят к уменьшению экскурсии грудной клетки и появлению вентиляционных нарушений по рестриктивному типу. В отличие от других САЗ, ПЛ при ПМ/ДМ не затрагивает в первую очередь дыхательные пути или плевру. Наиболее распространенной патологией является аспирационная пневмония, которая возникает по причине слабости мышц глотки и верхней трети пищевода.
Болезнь Шегрена
Болезнь Шегрена (БШ) — системное аутоиммунное заболевание неизвестной этиологии, характеризуется поражением секретирующих эпителиальных желез, с вовлечением преимущественно слюнных и слезных желез (ксеростомия, ксерофтальмия).
Поражение экзокринных желез верхних дыхательных путей часто приводит к сухости носовых ходов и бронхов. Наиболее распространенным проявлением ПЛ, связанным с БШ, является лимфоцитарный пневмонит, поражающий нижние доли. У пациентов с БШ может развиться плеврит (с или без выпота), ИПЛ с очагами лимфоидной инфильтрации. При развитии неходжкинских лимфом метастатические поражения легких встречаются часто, реже наблюдают формирование MALT-ткани (mucosal-associated lymphoid tissue) с развитием первичной MALT-лимфомы [1–3, 12, 13].
Смешанное заболевание соединительной ткани
Смешанное заболевание соединительной ткани (синдром Шарпа) (CЗСТ) — аутоиммунное заболевание, характеризующееся наличием отдельных признаков СКВ, ССД, РА, ПМ/ДМ в сочетании с высоким титром антител к экстрагируемому ядерному антигену — U1-RNP.
ИПЛ и ЛГ довольно часто встречаются у больных с СЗСТ, при этом нередко имеют субклиническое течение. Обследование пациентов с СЗСТ показало повышение уровня иммунных комплексов (ИК) и увеличение потребления комплемента. ИК-опосредованное повреждение альвеолярно-капиллярной мембраны и легочной ткани может играть важную роль в патогенезе ИПЛ при СЗСТ. При исследовании показателей функции внешнего дыхания больные с СЗСТ демонстрируют снижение диффузионной способности легких (ДСЛ) и рестриктивный тип нарушения вентиляции. Прогноз ИПЛ у пациентов с СЗСТ более благоприятный, чем при РА и ССД. ЛГ является основной причиной смерти больных с СЗСТ [1–3, 14, 15].
Гранулематоз Вегенера
Гранулематоз Вегенера (ГВ) — системный некротический васкулит мелких вен и артерий с образованием гранулем в сосудистых стенках и окружающих тканях дыхательных путей, почек и других органов.
ПЛ развивается у большинства пациентов с ГВ. Клинические проявления ПЛ при ГВ разнообразны, начиная от бессимптомных узелков в легких и кончая фульминантным альвеолярным кровотечением. ГВ может сопровождаться образованием опухолевидных инфильтратов с неровными краями, которые могут распадаться и образовывать полости. Плеврит, легочное кровотечение и увеличение лимфатических узлов средостения встречаются редко. Поражение трахеальных или бронхиальных стенок обычно проявляется гранулематозным утолщением слизистой оболочки или подслизистого слоя, при этом возникает обструктивный тип нарушения вентиляции легких. Частое осложнение — коллапс бронхов и постобструктивная пневмония. Инфильтраты, которые могут увеличиваться и уменьшаться, первоначально часто неправильно диагностируются как пневмония. Примерно в 20% случаев развивается прогрессирующая легочная недостаточность, связанная с ЛФ, пневмонией или пневмонитом, индуцированным циклофосфамидом. ДСЛ, как правило, уменьшена, но при развитии диффузных альвеолярных геморрагий наблюдают ее рост. Описаны случаи развития бронхоплевральных свищей [1–3].
Синдром Чарджа–Стросса
Синдром Чарджа–Строса (СЧС) — эозинофильное, гранулематозное воспаление респираторного тракта и некротизирующий васкулит, поражающий мелкие и средние сосуды, часто сочетающийся с астмой и эозинофилией.
Легкие — это наиболее поражаемый орган при данном синдроме; более 90% пациентов с синдромом СЧС в анамнезе имеют астму. При рентгенологическом исследовании легких выявляются очаги консолидации, распределяющиеся по периферии, которые часто бывают преходящими. Могут появляться узелки, при распаде не образующие полости. Другие менее распространенные проявления ПЛ включают утолщение междольковой перегородки и утолщение бронхиальной стенки. Плевральные выпоты образуются редко.
Существуют три фазы развития СЧС: продромальная фаза, которая характеризуется наличием аллергических заболеваний (как правило, астма или аллергический ринит), может продолжаться от нескольких месяцев до многих лет; эозинофилия/фаза инфильтрации тканей, в которой может наблюдаться удивительно высокая периферическая эозинофилия, а также инфильтрация эозинофилами тканей легких, желудочно-кишечного тракта и других органов; васкулитная фаза, в которой некротический васкулит поражает широкий спектр органов — сердце, легкие, периферические нервы и кожу. Диагноз приходится верифицировать с другими васкулитами, в первую очередь ГВ [1–3].
Синдром Гудпасчера
Синдром Гудпасчера (СГ) (геморрагический легочно-почечный синдром) — прогрессирующее аутоиммунное заболевание легких и почек, характеризующееся образованием антител к базальным мембранам капилляров клубочков почек и альвеол и проявляющееся сочетанием легочных и почечных геморрагий.
Патоморфологически в легких наблюдается картина венулитов, артериолитов, капилляритов с выраженными явлениями деструкции и пролиферации; поражение капилляров наблюдается преимущественно в межальвеолярных перегородках, развивается альвеолит с геморрагическим экссудатом в альвеолах.
В большинстве случаев ПЛ и почек происходит одновременно. Клинические проявления ПЛ включают в себя кашель, одышку и кровохарканье, которое может появляться на несколько месяцев раньше признаков поражения почек. В развитии альвеолита при СГ огромное значение имеет активация альвеолярных макрофагов. В активированном состоянии они выделяют около 40 цитокинов. Цитокины I группы (хемотаксины, лейкотриены, интерлейкин-8) усиливают поступление полиморфноядерных лейкоцитов в легкие. Цитокины II группы (факторы роста — тромбоцитарный, макрофагальный) способствуют перемещению в легкие фибробластов. Альвеолярные макрофаги продуцируют также активные формы кислорода, протеазы, повреждающие легочную ткань.
Исследование жидкости бронхоальвеолярного лаважа (ЖБАЛ) не является диагностическим при СГ, но может использоваться для подтверждения наличия диффузной альвеолярной геморрагии у пациентов с гломерулонефритом и легочными инфильтратами, но без кровохарканья. ЖБАЛ, которая остается геморрагической после многократных промываний, позволяет подтвердить диффузный геморрагический синдром, особенно при сопутствующем снижении гематокрита.
Гистологическое и иммунологическое исследование биоптатов легочной ткани при СГ характеризуется признаками геморрагического альвеолита, гемосидероза и интерстициального фиброза, а также линейных отложений иммуноглобулина G (IgG) и С3-компонента комплемента на базальных мембранах легочных альвеол.
Рентгено-компьютерное исследование легких при СГ демонстрирует наличие легочных инфильтратов в прикорневой области с распространением на нижние и средние отделы легких. Исследование легочных тестов выявляет рестриктивный тип нарушения вентиляции легких (снижение жизненной емкости легких — ЖЕЛ), но по мере прогрессирования заболевания присоединяются обструктивные изменения (снижение объема форсированного выдоха за 1 сек — ОФВ1, индекса Тиффно) [1–3].
Болезнь Бехтерева (анкилозирующий спондилит)
Болезнь Бехтерева (ББ, анкилозирующий спондилит, АС) — хроническое системное заболевание, характеризующееся воспалительным поражением суставов позвоночника, околопозвоночных тканей и крестцово-подвздошных сочленений с анкилозированием межпозвоночных суставов и развитием кальцификации спинальных связок.
ПЛ у больных АС встречается в 50–85% случаев и обусловлено анкилозирующим процессом в грудном отделе позвоночника, снижением дыхательной экскурсий грудной клетки, утомлением и слабостью дыхательных мышц. У больных АС чаще всего развивается эмфизема легких, затем ИПЛ, хроническая обструктивная болезнь легких (ХОБЛ), апикальный фиброз, бронхоэктазия и поражение плевры. Апикальный пневмофиброз, который встречается нечасто (3–4%), требует проведения дифференциальной диагностики с туберкулезными изменениями. Фиброз верхней доли легкого обычно протекает бессимптомно, но может вызывать кашель, отделение мокроты и одышку.
При АС чаще встречается рестриктивный тип нарушения вентиляции легких. У больных АС с хронической обструктивной болезнью легких (ХОБЛ) исследование ФВД демонстрирует обструктивные вентиляционные изменения [1, 16, 17].
Заключение
Таким образом, при САЗ могут наблюдаться различные типы легочной патологии. Развитие ПЛ обусловлено особенностями патофизиологических характеристик основного заболевания. Основные легочные проявления САЗ включают заболевания плевры, ИПЛ, поражение бронхиального дерева. При РА и СКВ чаще, чем при других САЗ, встречается поражение плевры. ИПЛ в настоящее время все больше признается как самое частое и серьезное проявление САЗ. ПЛ у больных с САЗ оказывает существенное негативное воздействие на качество жизни (КЖ): у больных снижаются показатели КЖ, характеризующие физический, психоэмоциональный статус и социальную активность.
ПЛ при САЗ имеет большое значение в формировании облика заболевания, при этом во многом определяет его тяжесть и прогноз. Наряду с базисной терапией САЗ, ПЛ необходимо рассматривать как важную мишень для терапевтического воздействия.
Литература
- Ревматология, национальное руководство. Под ред. Е. Л. Насонова, В. А. Насоновой. М.: ГЭОТАР-Медиа, 2008.
- Castelino F. V., Varga J. Interstitial lung disease in connective tissue diseases: evolving concepts of pathogenesis and management // Arthritis Research & Therapy. 2010; 12: 213.
- Bouros D., Pneumatikos I., Tzouvelekis A. Pleural involvement in systemic autoimmune disorders // Respiration. 2008; 75: 361–371.
- Kriegel M. A., Van Beek C., Mostaghimi A. et al. Sterile empyematous pleural effusion in a patient with systemic lupus erythematosus: a diagnostic challenge // Lupus. 2009; 18: 581–585.
- Pego-Reigosa J. M., Medeiros D. A., Osenberg D. A. Respiratory manifestations of systemic lupus erythematosus: old and new concepts // Best Pract Res Clin Rheumatol. 2009; 23: 460–480.
- Varda J., Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder // J Clin Invest. 2007; 117: 557–567.
- Yanaba K., Hasegawa M., Takehara K. et al. Comparative study of serum surfactant protein-D and KL-6 concentrations in patients with systemic sclerosis as markers for monitoring the activity of pulmonary fibrosis // J Rheumatol. 2004; 31: 1112–1120.
- McNearney T. A., Revelle J. D., Fischbach M. et al. Pulmonary involvement in systemic sclerosis: associations with genetic, serologic, sociodemographic, and behavioral factors // Arthritis Rheum. 2007; 57: 318–326.
- Tillie-Leblond I., Wislez M., Valeyre D. et al. Interstitial lung disease and anti-Jo-1 antibodies: difference between acute and gradual onset // Thorax. 2008; 63: 53–59.
- Chen I. J., Jan Wu Y. J., Lin C. W. et al. Interstitial lung disease in polymyositis and dermatomyositis // Clin Rheumatol. 2009; 28: 639–646.
- Fujisawa T., Suda T., Nakamura Y. et al. Differences in clinical features and prognosis of interstitial lung diseases between polymyositis and dermatomyositis // J Rheumatol. 2005; 32: 58–64.
- Ito I., Nagai S., Kitaichi M. et al. Pulmonary manifestations of primary Sjögren’s syndrome: a clinical, radiologic, and pathologic study // Am J Respir Crit Care Med. 2005; 171: 632–638.
- Parambil J. G., Myers J. L., Lindell R. M. et al. Interstitial lung disease in primary Sjögren’s syndrome // Chest. 2006; 130: 1489–1495.
- Bodolay E., Szekanecz Z., Devenyi K. et al. Evaluation of interstitial lung disease in mixed connective tissue disease (MCTD) // Rheumatol. 2005; 44: 656–661.
- Kinder B. W., Shariat C., Collard H. R. et al. Undifferentiated connective tissue disease-associated interstitial lung disease: changes in lung function // Lung 2010; 188: 143–149
- Quismorio F. P. Jr. Pulmonary involvement in ankylosing spondylitis // Curr Opin Pulm Med. 2006; 12: 342–345.
- Lee C. C., Lee S. H., Chang I. J. et al. Spontaneous pneumothorax associated with ankylosis spondylitis // Rheumatol. 2005; 44: 1538–1541.
Д. В. Бестаев 1 , кандидат медицинских наук
Е. Л. Насонов, доктор медицинских наук, профессор, академик РАН
Читайте также: