
Обмен фенилаланина и тирозина реферат
Обновлено: 05.07.2024
Автор текста – Анисимова Елена Сергеевна.
Авторские права защищены. Продавать текст нельзя.
Курсив не зубрить.
ПАРАГРАФ 68.1:
Обмен фенилаланина и тирозина
Содержание параграфа 68.1:
Повторение.
68.1.1. Превращение Фен в Тир
68.1.2. Синтез йодтиронинов из Тирозина
68.1.3. Синтез меланина из тирозина
68.1.4. Синтез катехоламинов из тирозина.
68.1.5. Катаболизм тирозина.
68.1.6. Тканевые особенности обмена Фен и Тир:
68.1.7. Метаболические блоки в обмене Тир и Фен.
Повторение сказанного в других параграфах:
1.
Фенилаланин и Тирозин являются белковыми АК,
и при их дефиците в организме синтез белков организма снижается;
причиной дефицита Фен и Тир в организме является их нехватка в пище (отсутствие полноценного белкового питания).
2.
Кроме белков, из Фенилаланина и Тирозина синтезируются
гормоны катехоламины (см. п. 63, 105)
и йодтиронины,
а также пигмент меланин.
3.
Из п.67:
Тирозин катаболизируется до метаболитов ЦТК
(фумарата и ацетилКоА),
которые могут превращаться в СО2,
в результате чего завершается катаболизм углеродного скелета Тир.
Этот путь метаболизма позволяет получить АТФ и тепло
при дефиците энергии,
а при дефиците глюкозы
из фумарата можно синтезировать глюкозу – п.66 и 55.
68.1.1. Превращение Фен в Тир
позволяет получить тирозин
и является реакцией синтеза Тир в организме.
Поскольку Фен в организме не синтезируется
(то есть является незаменимой АК),
то синтез Тир возможен при условии, что поступает Фен,
поэтому Тир относится к условно заменимым АК.
Но главное значение этой реакции не в получении тирозина,
т.к. Тирозин может поступать с пищей (в составе пищевых белков),
а в избавлении от Фенилаланина
и тем самым – в спасении от олигофрении (объяснения далее).
Фенилаланин нужен организму для синтеза белков,
но то количество Фенилаланина, которое не потратилось на синтез белков
(в том числе из-за дефицита других АК),
должно превратиться в Тирозин и метаболиты дальнейшего обмена Тир.
Иначе Фенилаланин накапливается
и превращается в побочные метаболиты своего обмена,
(в т.ч. фенилпируват в результате реакции пере/аминирования;
а также – фениллактат, фенилацетат),
накопление которых приводит к развитию ОЛИГОФРЕНИИ.
Фенилпируват выводится с мочой,
его присутствие в моче называется ФЕНИЛ/КЕТОН/УРИЕЙ.
(Перечитайте п.8).
Основная причина накопления Фенилаланина и его метаболитов
–
это низкая активность фермента,
превращающего Фенилаланин в Тирозин
(то есть Фенилаланин/гидроксилазы),
а основной причиной низкой активности этого фермента является дефект (мутация) гена,
кодирующего этого фермент
(то есть фенилпировиноградная олигофрения относится к первичным энзимопатиям,
наследственным заболеваниям, генетическим).
Развитие фенилпировиноградной олигофрении можно предотвратить
за счет специальной диеты:
нужно ограничить в питании количество продуктов,
содержащих Фенилаланин
(но не исключить совсем продукты с Фен из питания,
так как некоторое количество Фенилаланина нужно для синтеза белков при интенсивном росте детского организма) –
то есть принципом терапии
в этом случае первичной энзимопатии
является не генотерапия,
а ограничение поступления в организм субстрата реакции.
Ограничить нужно молоко
(в том числе материнское – за счёт перевода ребёнка на смеси с минимумом Фен),
мясо и другие белковые продукты.
Моча новорожденных в ряде стран тестируется на наличие фенилпирувата,
и в случае его обнаружения
ребенок питается специальными смесями
(молоко матери, как и многие другие продукты,
при фенилкетонурии ребенку противопоказано,
т.к. белки молока содержат Фенилаланин).
Здоровым надо знать:
дефицит витамина РР тоже снижает активность Фенилаланин/гидроксилазы
и (вследствие этого) приводит к накоплению Фенилаланина и его метаболитов,
это может способствовать ментальным нарушениям.
Авитаминоз РР называется пеллагрой,
одним из проявлений пеллагры является деменция (нарушение психики).
68.1.2. Синтез йодтиронинов из Тирозина
(См. п.104)
- происходит в щитовидной железе,
- требует стимуляции тиреотропином (гормонов гипофиза),
- наличия йода и здоровой ЩВЖ.
О последствиях избыточного и недостаточного синтеза ЙТ см. п.104.
68.1.3. Синтез меланина из тирозина
происходит в меланоцитах –
клетках, синтезирующих пигмент меланин.
Меланин синтезируется из ДОФА
(ДОФА является промежуточным метаболитом в синтезе и меланина, и катехоламинов,
то есть общим метаболитом этих синтезов – см. п.63).
Меланин выполняет функцию защиты от ультрафиолета;
при дефиците меланина кожа меньше защищена от ультрафиолета,
и загорать нужно более осторожно.
Иначе в лучшем случае кожа будет быстро стареть из-за загорания,
повышается риск рака кожи.
От интенсивности синтеза меланина зависит его количество,
а от количества меланина зависит:
1) насколько хорошо кожа защищена от ультрафиолета,
2) блондином или брюнетом будет человек,
3) насколько смуглой будет кожа,
4) карим или серым будет цвет глаз
(чем больше меланина, тем темнее все перечисленное – кожа, радужка).
68.1.4. Синтез катехоламинов из тирозина.
Кратко:
Катехоламины – это группа из трёх гормонов –
дофамин, норадреналин, адреналин.
Тирозин превращается в дофамин через ДОФА,
а из дофамина образуется адреналин через норадреналин.
Тирозин превращается в дофамин
в результате присоединения атома О и отщепления СО2.
После присоединения атома О тирозин становится ДОФА,
а после отщепления СО2 от ДОФА он становится дофамином.
Дофамин превращается в норадреналин
в результате присоединения атома кислорода (при гидросилировании)
к атому углерода в бывшем ;-положении.
Фермент – ДА-;-гидроксилаза,
нужен аскорбат в качестве источника 2Н,
поэтому при гиповитаминозе С синтез НА и А снижается.
Норадреналин превращается в адреналин
в результате присоединения СН3 (метильной) группы
к атому азота
путем переноса метила от SAM
(процесс называется (транс)метилированием,
фермент – норадреналин/метил/трансфераза).
О функциях катехоламинов см. 105.
Снижение синтеза катехоламинов приводит к снижению их функций и появлению ряда последствий.
68.1.5. Катаболизм тирозина.
При катаболизме тирозина образуются фумарат и ацетоацетат (бета-кетобутират).
Фумарат может использоваться для синтеза глюкозы
в ГНГ при голодании – п.67 и 33.
;-кетобутират – кетоновое тело, которое может питать все клетки
(кроме эритроцитов) при голодании – п.47.
В п.67 показывалось, что Фенилаланин превращается в Тирозин,
поэтому Фенилаланин может превращаться в те же вещества,
в которые превращается Тирозин.
Начинается катаболизм тирозина с реакции переаминирования (п.64):
тирозин вступает в реакцию с кетоглутаратом,
в результате КЕТОглутарат превращается в АМИНОглутаРат (=глутаМат),
а тирозин превращается в пара(4)-гидрокси/фенил/пируват:
вещество, у которого вместо аминогруппы тирозина есть кетогруппа.
Подсказка: тирозин – это гидроксифенилаланин.
Аланин превращается в пируват,
Фенил/АЛАНИН – в фенил/ПИРУВАТ,
Гидрокси/фенил/АЛАНИН (=тирозин) – в гидрокси/фенил/ПИРУВАТ.
Катализируется переаминирование
ферментами аминотрансферазами
с участием кофермента пиридоксальфосфата,
который образуется из пиридоксаля (витамина В6)
при присоединении к нему фосфата (при фосфорилировании).
4-гидрокси/фенил/пируват в ходе нескольких реакций
превращается в ГОМОГЕНТИЗИНОВУЮ кислоту (ГГК, гомогентизат),
при расщеплении которой образуются конечные метаболиты
катаболизма тирозина и фенилаланина:
фумарат и ацетоацетат.
При низкой активности фермента,
расщепляющего гомогентизиновую кислоту,
скорость расщепления этой кислоты снижается,
что приводит к накоплению гомогентизиновой кислоты и АЛКАПТОН/УРИИ.
При алкаптонурии повреждения организма менее серьёзные,
чем при фенилпировиноградной олигофрении,
но есть риск развития мочекаменной болезни.
Внешние проявления при алкаптонурии:
потемнение хрящей носа, ушей.
68.1.6. Тканевые особенности обмена Фен и Тир:
то есть в каких тканях (или клетках) во что они превращаются:
1) в клетках щитовидной железы – в гормоны ЙОДТИРОНИНЫ (п.104),
2) в мозговом веществе надпочечников – в гормон АДРЕНАЛИН,
3) в симпатических нервах и в головном мозге – в гормон НОРАДРЕНАЛИН,
4) в мозге, кишечнике, почках и сосудах – в гормон ДОФАМИН,
5) в меланоцитах – в пигмент МЕЛАНИН,
6) в печени – катаболизм до ФУМАРАТА и ацетоацетата.
Путь обмена Тир в конкретных клетках зависит от набора ферментов в этих клетках:
какие ферменты работают – такие реакции и протекают.
Например, ноадреналин/метил/трансфераза есть только в мозговом веществе надпочечников,
поэтому только здесь норадреналин может превратиться в адреналин,
поэтому только надпочечники производят и секретируют адреналин.
Набор ферментов в клетке зависит от того, какие гены работают (экспрессируются) в клетке,
то есть какие гены используются для синтеза РНК (транскрибируются),
какие РНК используются для синтеза ферментов и других белков. – п.84.
68.1.7. Метаболические блоки в обмене Тир и Фен.
Фенилкетонурия, альбинизм и алкаптонурия являются
примерами МЕТАБОЛИЧЕСКИХ БЛОКОВ и энзимопатий – п.8.
Метаболический блок – это снижение скорости реакции,
которое приводит к накоплению субстратов реакции
и дефициту продуктов реакции,
что может приводить к смерти или развитию болезни.
Причина метаболического блока
(сниженной скорости реакции) –
снижение активности фермента
- из-за мутации кодирующего гена
- или отсутствия кофактора
- из-за непоступления витамина с пищей
- или нарушения превращения витамина в кофактор
из-за дефицита ферментов активации витамина (п.11).
Фенилаланин - незаменимая аминокислота, так как в клетках животных не синтезируется её бензольное кольцо. Тирозин - условно заменимая аминокислота, поскольку образуется из фенилаланина. Содержание этих аминокислот в пищевых белках (в том числе и растительных) достаточно велико. Фенилаланин и тирозин используются для синтеза многих биологически
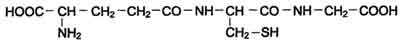
Схема ГГлутатион

Схема A

Схема Б
активных соединений. В разных тканях метаболизм этих аминокислот происходит поразному (рис. 9-28).
1. Метаболизм феиилаланина
Основное количество фенилаланина расходуется по 2 путям:
включается в белки;
превращается в тирозин.
Превращение фенилаланина в тирозин прежде всего необходимо для удаления избытка фенилаланина, так как высокие концентрации его токсичны для клеток. Образование тирозина не имеет большого значения, так как недостатка этой аминокислоты в клетках практически не бывает.
Основной путь метаболизма фенилаланина начинается с его гидроксилирования (рис. 9-29), в результате чего образуется тирозин. Эта реакция катализируется специфической монооксиге-назой - фенилаланингидр(жсилазой, кофермен-том которой служит тетрагидробиоптерин (Н4БП). Активность фермента зависит также от наличия Fe 2+ . Реакция необратима. Н4БП в результате реакции окисляется в дигидробиоптерин (Н2БП). Регенерация последнего происходит при участии дигидроптеридинредуктазы с использованием NADPH + H + .
2. Особенности обмена тирозина в разных тканях
Обмен тирозина значительно сложнее, чем обмен фенилаланина. Кроме использования в синтезе белков, тирозин в разных тканях выступает предшественником таких соединений, как катехоламины, тироксин, меланины, и ка-таболизируется до СО2 и Н2О.
Катаболизм тирозина в печени
В печени происходит катаболизм тирозина до конечных продуктов. Специфический путь катаболизма включает несколько ферментативных реакций, завершающихся образованием фумарата и ацетоацетата (см. схему А на с. 507):
Трансаминирование тирозина с ос-кетоглутаратом катализирует тирозинаминотрансфе-раза(кофермент ПФ) - индуцируемый фермент печени млекопитающих. В результате образуется п-гидроксифенилпируват.
В реакции окисления п-гидроксифенилпирувата в гомогентизиновую кислоту происходит декарбоксилирование, гидроксилирование ароматического кольца и миграция боковой цепи. Реакцию катализирует фермент п-гидроксифенилпируватдиоксигеназа, кофакторами которого выступают витамин С и Fe 2+ .
Превращение гомогентизиновой кислоты в фумарилацетоацетат сопровождается расщеплением ароматического кольца. Эта реакция катализируется диоксигеназой гомогентизиновой кислоты, в качестве кофермента содержащей Fe 2+ .
Обмен фенилаланина и тирозина связан со значительным количеством реакций гидроксилирования, которые катализируют оксигеназы. Ферменты оксигеназы (гидроксилазы) используют молекулу О2 и кофермент-донор водорода (чаще - Н4БП). Для катализа оксигеназам не-
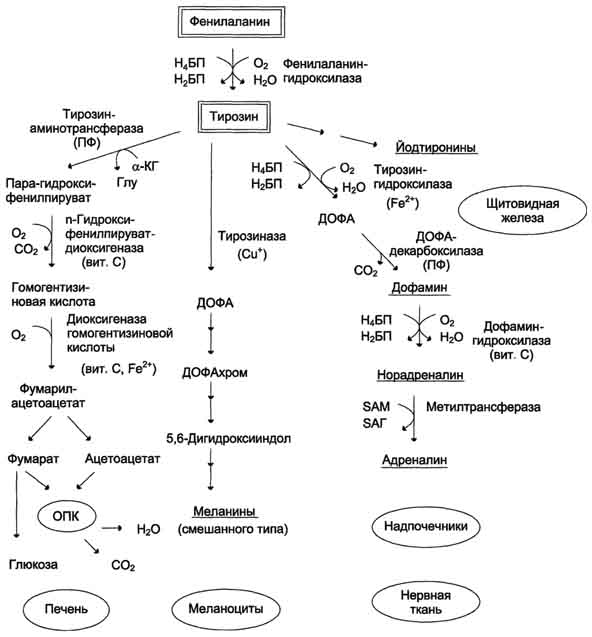
Рис. 9-28. Пути превращения фенилаланина и тирозина в разных тканях. Н4БП - тетрагидробиоптерин; Н2БП - дигидробиоптерин; ПФ - пиридоксальфосфат; SAM - S-аденозилметионин.
обходимы кофакторы - Fe 2+ или гем (для некоторых - Сu + ), а для многих ещё и витамин С. Оксигеназы делят на 2 группы:
Монооксигеназы - один атом О2 присоединяют к продукту реакции, другой используют для образования Н2О;
Диоксигеназы - оба атома О2 используют для образования продукта реакции.
Почти все процессы расщепления ароматических колец в биологических системах катализируются диоксигеназами, подклассом ферментов, открытым японским биохимиком Осами Хайяши.
В результате разрыва бензольного кольца образуется малеилацетоацетат, который в процессе цис- и транс-изомеризации превращается в фумарилацетоацетат.
Гидролиз фумарилацетоацетата при действии фумарилацетоацетатгидролазы приводит к образованию фумарата и ацетоацетата.
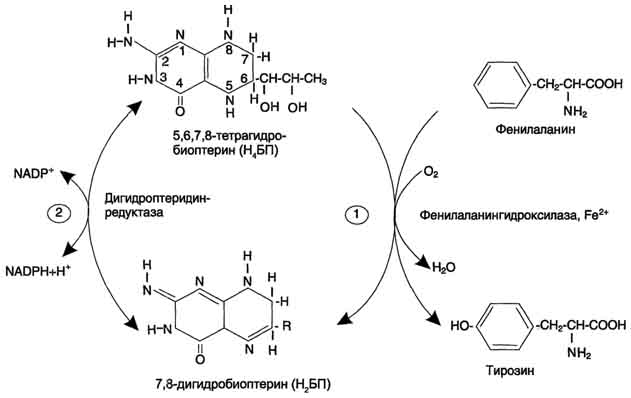
Рис. 9-29. Реакции гидроксилирования фенилаланина (1) и регенерации Н4БП (2).

Схема А
Фумарат может окисляться до СО2 и Н2О или использоваться для глюконеогенеза. Ацетоацетат - кетоновое тело, окисляемое до конечных продуктов с выделением энергии.
Превращение тирозина в меланоцитах
В пигментных клетках (меланоцитах) тирозин выступает предшественником тёмных пигментов - меланинов. Среди них преобладают 2 типа: эумеланины и феомеланины. Эумеланины (чёрного и коричневого цвета) - нерастворимые высокомолекулярные гетерополимеры 5,6-дигидроксииндола и некоторых его предшественников. Феомеланины - жёлтые или красновато-коричневые полимеры, растворимые в разбавленных щелочах. Находятся они, в основном, в составе волос. Меланины присутствуют в сетчатке глаз. Цвет кожи зависит от распределения меланоцитов и количества в них разных типов меланинов.
Синтез меланинов - сложный, многоступенчатый, разветвлённый процесс. Краткая схема синтеза представлена на рис. 9-28.
Первую реакцию - превращение тирозина в ДОФА - катализирует тирозиназа, использующая в качестве кофактора ионы Сu + (см. схему А на с. 509).
Превращение тирозина в щитовидной железе
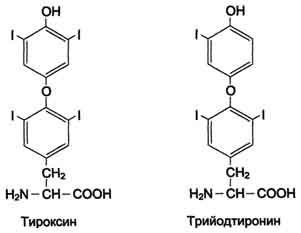
В щитовидной железе синтезируются и выделяются гормоны йодтиронины: тироксин (тет-райодтиронин) и трийодтиронин. Эти гормоны представляют собой йодированные остатки тирозина, которые попадают в клетки щитовидной железы через базальную мембрану (см. раздел 11).
Превращения тирозина в надпочечниках и нервной ткани (синтез катехоламинов)
В мозговом веществе надпочечников и нервной ткани тирозин является предшественником катехоламинов (дофамина, норадреналина и адреналина) (см. схему Б на с. 509).
При образовании катехоламинов, которое происходит в нервной ткани и надпочечниках, и меланина в меланоцитах промежуточным продуктом служит диоксифенилаланин (ДОФА) . Однако гидроксилирование тирозина в клетках различных типов катализируется различными ферментами:
Тирозиназа в меланоцитах является Сu + -зависимым ферментом (см. выше).
Тирозингидроксилаза (1) в надпочечниках и ка-техоламинергических нейронах не нуждается в ионах меди. Это - Fе 2+ -зависимый фермент, аналогично фенилаланингидроксилазе в качестве кофермента использующий Н4БП.
Физиологическая роль тирозингидроксилазы чрезвычайно велика, так как этот фермент является регуляторным и определяет скорость синтеза катехоламинов.
Активность тирозингидроксилазы значительно изменяется в результате:
Аллостерической регуляции (ингибитор - норадреналин);
Фосфорилирования/дефосфорилирования: в результате фосфорилирования с участием протеинкиназы А снижаются Кm для кофермента Н4БП и сродство фермента к норадреналину, в результате чего происходит активация тирозингидроксилазы.
Количество фермента регулируется на уровне транскрипции.
ДОФА-декарбоксилаза (2) (кофермент - ПФ) катализирует образование дофамина, который при участии дофамингидроксилазы (3) (монооксигеназы) превращается в норадреналин. Для функционирования фермента необходимы ионы Сu + , витамин С и тетрагидробиоптерин.
В мозговом веществе надпочечников фенилэтаноламин-N-метилтрансфераза (4) катализирует метилирование норадреналина, в результате чего образуется адреналин. Источником метальной группы служит &АМ.
Дофамин и норадреналин служат медиаторами в синаптической передаче нервных импульсов,

Схема А

Схема Б
а адреналин - гормон широкого спектра действия, регулирующий энергетический обмен. Одна из функций катехоламинов - регуляция деятельности ССС (см. раздел 11).
3. Заболевания, связанные с нарушением обмена фенилаланина и тирозина
Известно несколько наследственных заболеваний, связанных с дефектом ферментов обмена фенилаланина и тирозина в разных тканях.
Фенилкетонурия
В печени здоровых людей небольшая часть фенилаланина (∼10%) превращается в фенил-лактат и фенилацетилглутамин (рис. 9-30).
Этот путь катаболизма фенилаланина становится главным при нарушении основного пути - превращения в тирозин, катализируемого фенил-аланингидроксилазой. Такое нарушение сопровождается гиперфенилаланинемией и повышением в крови и моче содержания метаболитов
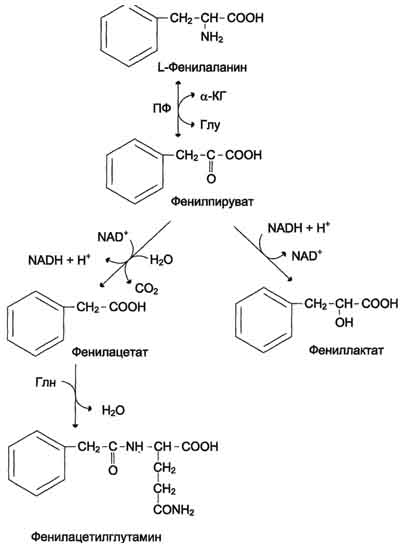
Рис. 9-30. Альтернативные пути катаболизма фенилаланина. При дефекте фенилаланингидроксилазы накопившийся фенилалан и н подвергается трансаминированию с а-кетоглутаратом. Образовавшийся фенилпируват превращается либо в фениллактат, либо в фенилацетилглутамин, которые накапливаются в крови и выделяются с мочой. Эти соединения токсичны для клеток мозга.
альтернативного пути: фенилпирувата, фенилацетата, фениллактата и фенилацетилглу-тамина. Дефект фенилаланингидроксилазы приводит к заболеванию фенилкетонурия (ФКУ). Выделяют 2 формы ФКУ:
Классическая ФКУ - наследственное заболевание, связанное с мутациями в гене фенилаланингидроксилазы, которые приводят к снижению активности фермента или полной его инактивации. При этом концентрация фенилаланина повышается в крови в 20-30 раз (в норме - 1,0-2,0 мг/дл), в моче - в 100-300 раз по сравнению с нормой (30 мг/дл). Концентрация фенилпирувата и фениллактата в моче достигает 300-600 мг/дл при полном отсутствии в норме.
Наиболее тяжёлые проявления ФКУ - нарушение умственного и физического развития, судорожный синдром, нарушение пигментации. При отсутствии лечения больные не доживают до 30 лет. Частота заболевания - 1:10 000 новорождённых. Заболевание наследуется по аутосомно-рецессивному типу.
Тяжёлые проявления ФКУ связаны с токсическим действием на клетки мозга высоких концентраций фенилаланина, фенилпирувата, фениллактата. Большие концентрации фенилаланина ограничивают транспорт тирозина
и триптофана через гематоэнцефаличеекий барьер и тормозят синтез нейро-медиаторов (дофамина, норадреналина, серотонина).
Вариантная ФКУ (коферментзависимая гиперфенилаланинемия) - следствие мутаций в генах, контролирующих метаболизм Н4БП. Клинические проявления - близкие, но не точно совпадающие с проявлениями классической ФКУ. Частота заболевания - 1-2 случая на 1 млн новорождённых.
Н4БП необходим для реакций гидроксилирования не только фенилаланина, но также тирозина и триптофана, поэтому при недостатке этого кофермента нарушается метаболизм всех 3 аминокислот, в том числе и синтез ней-ромедиаторов. Заболевание характеризуется тяжёлыми неврологическими нарушениями и ранней смертью ("злокачественная" ФКУ).
Прогрессирующее нарушение умственного и физического развития у детей, больных ФКУ, можно предотвратить диетой с очень низким содержанием или полным исключением фенилаланина. Если такое лечение начато сразу после рождения ребёнка, то повреждение мозга предотвращается. Считается, что ограничения в питании могут быть ослаблены после 10-летнего возраста (окончание процессов миелиниза-ции мозга), однако в настоящее время многие педиатры склоняются в сторону "пожизненной диеты".
Для диагностики ФКУ используют качественные и количественные методы обнаружения патологических метаболитов в моче, определение концентрации фенилаланина в крови и моче. Дефектный ген, ответственный за фенилкетонурию, можно обнаружить у фенотипически нормальных гетерозиготных носителей с помощью теста толерантности к фенилаланину. Для этого обследуемому дают натощак ∼10 г фенилаланина в виде раствора, затем через часовые интервалы берут пробы крови, в которых определяют содержание тирозина. В норме концентрация тирозина в крови после фенилаланиновой нагрузки значительно выше, чем у гетерозиготных носителей гена фежилкетонурии. Этот тест используется в генетической консультации для определения риска рождения больного ребёнка. Разработана схема скрининга для выявления новорождённых детей с ФКУ. Чувствительность теста практически достигает 100%.
В настоящее время диагностику мутантного гена, ответственного за ФКУ, можно проводить с помощью методов ДНК-диагностики (рестрикционного анализа и ПЦР).
Тирозинемии
Некоторые нарушения катаболизма тирозина в печени приводят к тирозинемии и тирози-нурии. Различают 3 типа тирозинемии.
Тирозинемия типа 1 (тирозиноз). Причиной заболевания является, вероятно, дефект фермента фумарилацетоацетатгидролазы, катализирующего расщепление фумарилацетоа-цетата на фумарат и ацетоацетат (рис. 9-28). Накапливающиеся метаболиты снижают активность некоторых ферментов и транспортных систем аминокислот. Патофизиология этого нарушения достаточно сложна. Острая форма тирозиноза характерна для новорождённых. Клинические проявления - диарея, рвота, задержка в развитии. Без лечения дети погибают в возрасте 6-8 мес из-за развивающейся недостаточности печени.Хроническая форма характеризуется сходными, но менее выраженными симптомами. Гибель наступает в возрасте 10 лет. Содержание тирозина в крови у больных в несколько раз превышает норму. Для лечения используют диету с пониженным содержанием тирозина и фенилаланина.
Тирозинемия типа II (синдром Рихнера-Ханхорта). Причина - дефект фермента тирозина-минотрансферазы. Концентрация тирозина в крови больных повышена. Для заболевания характерны поражения глаз и кожи, умеренная умственная отсталость, нарушение координации движений.
Тирозинемия новорождённых (кратковременная). Заболевание возникает в результате снижения активности фермента п-гидроксифенилпируватдиоксигеназы, превращающего п-гидроксифенилпируват в гомогентизиновую кислоту (рис. 9-28). В результате в крови больных повышается концентрация п-гидроксифенилацетата, тирозина и фенил-аланина. При лечении назначают бедную белком диету и витамин С.
Алкаптонурия ("чёрная моча")
Причина заболевания - дефект диоксигеназы гомогентизиновой кислоты (рис. 9-28). Для
этой болезни характерно выделение с мочой большого количества гомогентизиновой кислоты, которая, окисляясь кислородом воздуха, образует тёмные пигменты алкаптоны. Это метаболическое нарушение было описано ещё в XVI веке, а само заболевание охарактеризовано в 1859 г. Клиническими проявлениями болезни, кроме потемнения мочи на воздухе, являются пигментация соединительной ткани (охроноз) и артрит. Частота - 2-5 случаев на 1 млн новорождённых. Заболевание наследуется по аутосомнорецессивному типу. Диагностических методов выявления гетерозиготных носителей дефектного гена к настоящему времени не найдено.
Альбинизм
Причина метаболического нарушения - врождённый дефект тирозиназы. Этот фермент катализирует превращение тирозина в ДОФА в меланоцитах. В результате дефекта тирозиназы нарушается синтез пигментов меланинов.
Клиническое проявление альбинизма (от лат. albus - белый) - отсутствие пигментации кожи и волос. У больных часто снижена острота зрения, возникает светобоязнь. Длительное пребывание таких больных под открытым солнцем приводит к раку кожи. Частота заболевания 1:20 000.
Нарушение синтеза катехоламинов (рис. 9-28) может вызывать различные нервно-психические заболевания, причём патологические отклонения наблюдаются как при снижении, так и при увеличении их количества.
Болезнь Паркинсона
Заболевание развивается при недостаточности дофамина в чёрной субстанции мозга. Это одно из самых распространённых неврологических заболеваний (частота 1:200 среди людей старше 60 лет). При этой патологии снижена активность тирозингидроксилазы, ДОФА-декарбоксилазы. Заболевание сопровождается тремя основными симптомами: акинезия (скованность движений), ригидность (напряжение мышц), тремор (непроизвольное дрожание). Дофамин не проникает через гематоэнцефалический барьер и как лекарственный препарат не используется. Для лечения паркинсонизма предлагаются следующие принципы:
заместительная терапия препаратами-предшественниками дофамина (производными ДОФА) - леводопа, мадопар, наком и др.
подавление инактивации дофамина ингибиторами МАО (депренил, ниаламид, пиразидол и др.).
Депрессивные состояния часто связаны со снижением в нервных клетках содержания дофамина и норадреналина.
Гиперсекреция дофамина в височной доле мозга наблюдается при шизофрении.
Фенилаланин — незаменимая аминокислота, так как в клетках животных не синтезируется её бензольное кольцо. Тирозин — условно заменимая аминокислота, поскольку образуется из фенилаланина. Содержание этих аминокислот в пищевых белках (в том числе и растительных) достаточно велико. Фенилаланин и тирозин используются для синтеза многих биологически активных соединений. В разных тканях метаболизм этих аминокислот происходит по-разному (рис. 9-28).

Рис. 9-28. Пути превращения фенилаланина и тирозина в разных тканях. Н4ВП — тетрагидробиоптерин; Н2ВП — дигидроби-оптерин; ПФ — пиридоксальфосфат; SAM — S-аденозил метионин
1. Метаболизм фенилаланина
- включается в белки;
- превращается в тирозин.
Превращение фенилаланина в тирозин прежде всего необходимо для удаления избытка фенилаланина, так как высокие концентрации его токсичны для клеток. Образование тирозина не имеет большого значения, так как недостатка этой аминокислоты в клетках практически не бывает.
Основной путь метаболизма фенилаланина начинается с его гидроксилирования (рис. 9-29), в результате чего образуется тирозин. Эта реакция катализируется специфической монооксигеназой — фенилаланингидроксилазой, кофермен-том которой служит тетрагидробиоптерин (Н4БП). Активность фермента зависит также от наличия Fe 2+ . Реакция необратима. Н4БП в результате реакции окисляется в дигидробиоптерин (Н2БП). Регенерация последнего происходит при участии дигидроптеридинредуктазы с использованием NADPH + Н + .

Рис. 9-29. Реакции гидроксилирования фенилаланина (1) и регенерации Н4БП (2).
2. Особенности обмена тирозина в разных тканях
Обмен тирозина значительно сложнее, чем обмен фенилаланина. Кроме использования в синтезе белков, тирозин в разных тканях выступает предшественником таких соединений, как катехоламины, тироксин, меланины, и ка-таболизируется до СО2и Н2О.
Катаболизм тирозина в печени
печени происходит катаболизм тирозина до конечных продуктов. Специфический путь катаболизма включает несколько ферментативных реакций, завершающихся образованием фумарата и ацетоацетата (см. схему А ):
Схема А

1. Трансаминирование тирозина с а-кетоглутаратом катализирует тирозинаминотрансфераза (кофермент ПФ) — индуцируемый фермент печени млекопитающих. В реультате образуется п-гидроксифенилпируват.
2. В реакции окисления п-гидроксифенилпи-рувата в гомогентизиновую кислоту происходит декарбоксилирование, гидроксилирование ароматического кольца и миграция боковой цепи. Реакцию катализирует фермент п-гид-роксифенилпируватдиоксигеназа, кофакторами которого выступают витамин С и Fe 2+ .
3. Превращение гомогентизиновой кислоты в фумарилацетоацетат сопровождается расщеплением ароматического кольца. Эта реакция катализируется диоксигеназой гомогентизиновой кислоты, в качестве кофермента содержащей Fe 2+ .
- Монооксигеназы — один атом O2 присоединяют к продукту реакции, другой используют для образования Н2О;
- Диоксигеназы — оба атома O2 используют для образования продукта реакции.
Почти все процессы расщепления ароматических колец в биологических системах катализируются диоксигеназами, подклассом ферментов, открытым японским биохимиком Осами Хайяши.
В результате разрыва бензольного кольца образуется малеилацетоацетат, который в процессе цис- и транс-изомеризации превращается в фумарил ацетоацетат.
4. Гидролиз фумарил ацетоацетата при действии фумарилацетоацетатгидролазы приводит к образованию фумарата и ацетоацетата.
Фумарат может окисляться до СО2и Н2О или использоваться для глюконеогенеза. Ацетоацетат — кетоновое тело, окисляемое до конечных продуктов с выделением энергии.
Превращение тирозина в меланоцитах
В пигментных клетках (меланоцитах) тирозин выступает предшественником тёмных пигментов — меланинов. Среди них преобладают 2 типа: эумеланины и феомеланины. Эумелани-ны (чёрного и коричневого цвета) — нерастворимые высокомолекулярные гетерополимеры 5,6-дигидроксииндола и некоторых его предшественников. Феомеланины — жёлтые или красновато-коричневые полимеры, растворимые в разбавленных щелочах. Находятся они, в основном, в составе волос. Меланины присутствуют в сетчатке глаз. Цвет кожи зависит от распределения меланоцитов и количества в них разных типов меланинов.
Синтез меланинов — сложный, многоступенчатый, разветвлённый процесс. Краткая схема синтеза представлена на рис. 9-28.
Первую реакцию — превращение тирозина в ДОФА — катализирует тирозиназа, использующая в качестве кофактора ионы Сu + (см. схему Б).
Схема Б

Превращение тирозина в щитовидной железе
В щитовидной железе синтезируются и выделяются гормоны йодтиронины: тироксин (тетрайодтиронин) и трийодтиронин. Эти гормоны представляют собой йодированные остатки тирозина, которые попадают в клетки щитовидной железы через базальную мембрану (см. раздел 11).

Превращения тирозина в надпочечниках и нервной ткани (синтез катехоламинов)
В мозговом веществе надпочечников и нервной ткани тирозин является предшественником катехоламинов (дофамина, норадреналина и адреналина) (см. схему В).
Схема В

При образовании катехоламинов, которое происходит в нервной ткани и надпочечниках, и меланина в меланоцитах промежуточным продуктом служит диоксифенилаланин (ДОФА) . Однако гидроксилирование тирозина в клетках различных типов катализируется различными ферментами:
Тирозиназа в меланоцитах является Сu + -зависимым ферментом (см. выше). Тирозингвдроксилаза (1) в надпочечниках и ка-техоламинергических нейронах не нуждается в ионах меди. Это — Fe 2+ -зависимый фермент, аналогично фенил ал анингидроксилазе в качестве кофермента использующий Н4БП. Физиологическая роль тирозингидрокси-лазы чрезвычайно велика, так как этот фермент является регуляторным и определяет скорость синтеза катехоламинов. Активность тирозингидроксилазы значительно изменяется в результате: Аллостерической регуляции (ингибитор — норадреналин); Фосфорилирования/дефосфорилирования: в результате фосфорилирования с участием протеинкиназы А снижаются Кm для кофермента Н4БП и сродство фермента к норадреналину, в результате чего происходит активация тирозингидроксилазы. Количество фермента регулируется на уровне транскрипции.
ДОФА-декарбоксилаза (2) (кофермент — ПФ) катализирует образование дофамина, который при участии дофаминщдроксилазы (3) (монооксигеназы) превращается в норадреналин. Для функционирования фермента необходимы ионы Сu + , витамин С и тетрагидробиоптерин.
В мозговом веществе надпочечников фенил-этанол амин- N-метилтрансфераза (4) катализирует метилирование норадреналина, в результате чего образуется адреналин. Источником метильной группы служит SAM. Дофамин и норадреналин служат медиаторами в синаптической передаче нервных импульсов, а адреналин — гормон широкого спектра действия, регулирующий энергетический обмен. Одна из функций катехоламинов — регуляция деятельности ССС (см. раздел 11). 3. Заболевания, связанные с нарушением обмена фенилаланина и тирозина
Известно несколько наследственных заболеваний, связанных с дефектом ферментов обмена фенилаланина и тирозина в разных тканях.
Фенилкетонурия В
печени здоровых людей небольшая часть фенилаланина (~10%) превращается в фениллактат и фенилацетилглутамин (рис. 9-30).

Рис. 9-30. Альтернативные пути катаболизма фенилаланина. При дефекте фенилаланингидроксилазы накопившийся фенилаланин подвергается трансаминированию с α -кетоглутаратом. Образовавшийся фенил пируват превращается либо в фениллактат, либо в фенилацетилглутамин, которые накапливаются в крови и выделяются с мочой. Эти соединения токсичны для клеток мозга
Этот путь катаболизма фенилаланина становится главным при нарушении основного пути — превращения в тирозин, катализируемого фенил-аланингидроксилазой. Такое нарушение сопровождается гиперфенилаланинемией и повышением в крови и моче содержания метаболитов альтернативного пути: фенилпирувата, фенилацетата, фениллактата и фенилацетилглутамина. Дефект фенилаланингидроксилазы приводит к заболеванию фенилкетонурия (ФКУ). Выделяют 2 формы ФКУ:
Классическая ФКУ — наследственное заболевание, связанное с мутациями в гене фенила-ланингидроксилазы, которые приводят к снижению активности фермента или полной его инактивации. При этом концентрация фенилаланина повышается в крови в 20—30 раз (в норме — 1,0—2,0 мг/дл), в моче — в 100—300 раз по сравнению с нормой (30 мг/дл). Концентрация фенилпирувата и фениллактата в моче достигает 300—600 мг/дл при полном отсутствии в норме.
Наиболее тяжёлые проявления ФКУ — нарушение умственного и физического развития, судорожный синдром, нарушение пигментации. При отсутствии лечения больные не доживают до 30 лет. Частота заболевания — 1:10 000 новорождённых. Заболевание наследуется по аутосомно-рецессивному типу.
Тяжёлые проявления ФКУ связаны с токсическим действием на клетки мозга высоких концентраций фенилаланина, фенилпирувата, фениллактата. Большие концентрации фенилаланина ограничивают транспорт тирозина и триптофана через гематоэнцефалический барьер и тормозят синтез нейро-медиаторов (дофамина, норадреналина, серотонина).
Вариантная ФКУ (коферментзависимая ги-перфенилаланинемия) — следствие мутаций в генах, контролирующих метаболизм Н4БП. Клинические проявления — близкие, но не точно совпадающие с проявлениями классической ФКУ. Частота заболевания — 1—2 случая на 1 млн новорождённых.
Для диагностики ФКУ используют качественные и количественные методы обнаружения патологических метаболитов в моче, определение концентрации фенилаланина в крови и моче. Дефектный ген, ответственный за фенилкетону-рию, можно обнаружить у фенотипически нормальных гетерозиготных носителей с помощью теста толерантности к фенилаланину. Для этого обследуемому дают натощак ~10 г фенилаланина в виде раствора, затем через часовые интервалы берут пробы крови, в которых определяют содержание тирозина. В норме концентрация тирозина в крови после фенилаланиновой нагрузки значительно выше, чем у гетерозиготных носителей гена фенилкетонурии. Этот тест используется в генетической консультации для определения риска рождения больного ребёнка. Разработана схема скрининга для выявления новорождённых детей с ФКУ. Чувствительность теста практически достигает 100%.
В настоящее время диагностику мутантного гена, ответственного за ФКУ, можно проводить с помощью методов ДНК-диагностики (рестрикционного анализа и ПЦР).
Тирозинемии
Некоторые нарушения катаболизма тирозина в печени приводят к тирозинемии и тирози-нурии. Различают 3 типа тирозинемии.
Тирозинемия типа 1 (тирозиноз). Причиной заболевания является, вероятно, дефект фермента фумарилацетоацетатгидролазы, катализирующего расщепление фумарилацетоа-цетата на фумарат и ацетоацетат (рис. 9-28). Накапливающиеся метаболиты снижают активность некоторых ферментов и транспортных систем аминокислот. Патофизиология этого нарушения достаточно сложна. Острая форма тирозиноза характерна для новорождённых. Клинические проявления — диарея, рвота, задержка в развитии. Без лечения дети погибают в возрасте 6—8 мес из-за развивающейся недостаточности печени. Хроническая форма характеризуется сходными, но менее выраженными симптомами. Гибель наступает в возрасте 10 лет. Содержание тирозина в крови у больных в несколько раз превышает норму. Для лечения используют диету с пониженным содержанием тирозина и фенилаланина.
Тирозинемия типа II (синдром Рихнера—Ханхор-та). Причина — дефект фермента тирозина-минотрансферазы. Концентрация тирозина в крови больных повышена. Для заболевания характерны поражения глаз и кожи, умеренная умственная отсталость, нарушение координации движений.
Тирозинемия новорождённых (кратковременная). Заболевание возникает в результате снижения активности фермента п - гидро ксифенил-пируватдиоксигеназы, превращающего n-гидроксифенилпируват в гомогентизиновую кислоту (рис. 9-28). В результате в крови больных повышается концентрация п-гидрокси-фенилацетата, тирозина и фенил-аланина. При лечении назначают бедную белком диету и витамин С.
Причина заболевания — дефект диоксигеназы гомогентизиновой кислоты (рис. 9-28). Для этой болезни характерно выделение с мочой большого количества гомогентизиновой кислоты, которая, окисляясь кислородом воздуха, образует тёмные пигменты алкаптоны. Это метаболическое нарушение было описано ещё в XVI веке, а само заболевание охарактеризовано в 1859 г. Клиническими проявлениями болезни, кроме потемнения мочи на воздухе, являются пигментация соединительной ткани (охроноз) и артрит. Частота — 2—5 случаев на 1 млн новорождённых. Заболевание наследуется по аутосомно-рецессивному типу. Диагностических методов выявления гетерозиготных носителей дефектного гена к настоящему времени не найдено.
Альбинизм
Причина метаболического нарушения — врождённый дефект тирозиназы. Этот фермент катализирует превращение тирозина в ДОФА в меланоцитах. В результате дефекта тирозиназы нарушается синтез пигментов меланинов.
Клиническое проявление альбинизма (от лат. albus — белый) — отсутствие пигментации кожи и волос. У больных часто снижена острота зрения, возникает светобоязнь. Длительное пребывание таких больных под открытым солнцем приводит к раку кожи. Частота заболевания 1:20 000
Нарушение синтеза катехоламинов (рис. 9-28) может вызывать различные нервно-психические заболевания, причём патологические отклонения наблюдаются как при снижении, так и при увеличении их количества.
Болезнь Паркинсона
- аместительная терапия препаратами-пред-шественниками дофамина (производными ДОФА) — леводопа, мадопар, наком и др.
- подавление инактивации дофамина ингибиторами МАО (депренил, ниаламид, пиразидол и др.).
Депрессивные состояния часто связаны со снижением в нервных клетках содержания дофамина и норадреналина.
Фенилкетонурия (ФКУ) — генетическое заболевание, характеризующееся нарушениями обмена фенилаланина. Встречается с частотой 1 на 8000–15 000 новорожденных. Выделяют четыре формы ФКУ; существует свыше 400 различных мутаций и несколько метаболических фенотип
.jpg)
Фенилкетонурия (ФКУ) — генетическое заболевание, характеризующееся нарушениями обмена фенилаланина. Встречается с частотой 1 на 8000–15 000 новорожденных. Выделяют четыре формы ФКУ; существует свыше 400 различных мутаций и несколько метаболических фенотипов ФКУ [1].
Определение, патогенез, классификация
Фенилкетонурия — наследственная аминоацидопатия, связанная с нарушением метаболизма фенилаланина, в результате мутационной блокады ферментов приводящая к стойкой хронической интоксикации и поражению ЦНС c выраженным снижением интеллекта и неврологическим дефицитом [1, 2].
Основное значение в патогенезе классической ФКУ имеет неспособность фенилаланингидроксилазы перерабатывать фенилаланин до тирозина. В результате в организме накапливается фенилаланин и продукты его аномального обмена (фенилпировиноградная, фенилуксусная, фенилмолочная кислоты) [1–3].
В числе других патогенетических факторов рассматриваются нарушения аминокислотного транспорта через гематоэнцефалический барьер, нарушения церебрального пула аминокислот с последующим нарушением синтеза протеолипидных белков, нарушения миелинизации, низкие уровни нейротрансмиттеров (серотонин и др.) [1–4].
Фенилкетонурия I (классическая или тяжелая) — аутосомно-рецессивное заболевание, вызванное мутацией гена фенилаланингидроксилазы (длинное плечо хромосомы 12); выявлены 12 различных гаплотипов, из которых около 90% ФКУ ассоциировано с четырьмя гаплотипами. Наиболее частые мутации в гене фенилаланингидроксилазы: R408W, R261Q, IVS10 nt 546, Y414C. В основе болезни — дефицит фенилаланин-4-гидроксилазы, обеспечивающей конверсию фенилаланина в тирозин, что приводит к накоплению в тканях и физиологических жидкостях фенилаланина и его метаболитов [1–4].
Особую группу составляют атипичные варианты ФКУ, при которых клиническая картина напоминает классическую форму болезни, но по показателям развития, несмотря на проведение диетотерапии, не отмечается положительной динамики. Эти варианты ФКУ связаны с дефицитом тетрагидроптерина, дегидроптеринредуктазы, 6-пирувоилтетрагидроптеринсинтазы, гуанозин-5-трифосфатциклогидролазы и т. д. [1–4].
Фенилкетонурия II (атипичная) — аутосомно-рецессивное заболевание, при котором генный дефект локализуется в коротком плече хромосомы 4 (участок 4р15.3), характеризующееся недостаточностью дегидроптеринредуктазы, приводящей к нарушению восстановления активной формы тетрагидробиоптерина (кофактор в гидроксилировании фенилаланина, тирозина и триптофана) в сочетании со снижением в сыворотке крови и спинномозговой жидкости фолатов. Результатом являются метаболические блоки в механизмах превращения фенилаланина в тирозин, а также предшественников нейромедиаторов катехоламинового и серотонинового рядов (L-дофа, 5-окситриптофан). Болезнь описана в 1974 г. [1–4].
Фенилкетонурия III (атипичная) — аутосомно-рецессивное заболевание, связанное с недостаточностью 6-пирувоилтетрагидроптеринсинтазы, участвующей в процессе синтеза тетрагидробиоптерина из дигидронеоптеринтрифосфата (описано в 1978 г.). Дефицит тетрагидробиоптерина приводит к расстройствам, сходным с нарушениями при ФКУ II [1–4].
Примаптеринурия — атипичная ФКУ у детей с легкой гиперфенилаланинемией, у которых в моче в больших количествах присутствует примаптерин и некоторые его производные при наличии нормальной концентрации в спинномозговой жидкости нейромедиаторных метаболитов (гомованилиновой и 5-оксииндолуксусной кислот). Энзиматический дефект пока не выявлен [1–4].
Материнская ФКУ — заболевание, сопровождающееся снижением уровня интеллекта (до умственной отсталости) среди потомства женщин, страдающих ФКУ и не получающих специализированную диету в совершеннолетнем возрасте. Патогенез материнской ФКУ детально не изучен, но предполагается ведущая роль хронической интоксикации плода фенилаланином и продуктами его аномального метаболизма [1–4].
R. Koch и соавт. (2008) при аутопсии головного мозга младенца, у матери которого отмечалась ФКУ (без адекватного контроля за уровнем фенилаланина в крови), обнаружили ряд патологических изменений: низкий вес мозга, вентикуломегалию, гипоплазию белого вещества и задержку миелинизации (без признаков астроцитоза); хронических изменений в сером веществе головного мозга не было обнаружено. Предполагается, что нарушения в развитии белого вещества мозга ответственны за формирование неврологического дефицита при материнской ФКУ [5].
В практических целях в медико-генетических центрах РФ используется условная классификация ФКУ, основанная на уровнях содержания фенилаланина в сыворотке крови: классическая (тяжелая или типичная) — уровень фенилаланина выше 20 мг% (1200 мкмоль/л); средняя — 10,1–20 мг% (600–1200 мкмоль/л), а также уровень фенилаланина 8,1–10 мг%, если он устойчив на фоне физиологической нормы потребления белка в рационе питания; легкая (гиперфенилаланинемия, не требующая лечения) — уровень фенилаланина до 8 мг% (480 мкмоль/л) [2].
Клинические проявления и диагностика
При рождении дети с ФКУ I выглядят здоровыми, хотя чаще имеется специфический хабитус (светлые волосы, голубые глаза, суховатая кожа). При отсутствии своевременного выявления и лечения болезни в течение первых двух месяцев жизни у них появляется частая и интенсивная рвота и повышенная раздражительность. Между 4 и 9 месяцами становится очевидным выраженное отставание в психомоторном развитии [1–4].
В. М. Студеникин, доктор медицинских наук, профессор
Т. Э. Боровик, доктор медицинских наук, профессор
Т. В. Бушуева, кандидат медицинских наук
Читайте также: