
Кинетика ферментативных реакций реферат
Обновлено: 05.07.2024
Ферментативный катализ существенно отличается от неферментативного, в связи с чем в кинетике ферментативных реакций разработаны совершенно особые закономерности. Они позволяют выделить ферментативную кинетику в самостоятельный раздел химической кинетики, в котором изучается зависимость скорости реакций, катализируемых ферментами, от концентрации реагирующих веществ (ферментов и субстратов) и от условий их взаимодействия (температуры, рН, концентрации
коферментов и кофакторов, наличия различных эффекторов: активаторов и ингибиторов).
Изучение кинетики ферментативного действия имеет важное теоретическое значение, поскольку только с позиций кинетики можно подойти к решению вопроса о механизме ферментативного действия. Но оно также необходимо с практических позиций, так как только имея определенные сведения о кинетике действия того или иного фермента, можно подобрать оптимальные условия для его работы, а также влиять на его активность в заданном направлении на различных стадиях технологического процесса.
Вопросы, связанные с кинетикой ферментативных реакций, детально изложены в специальных разделах биохимии и энзимологии, поэтому основное внимание уделим тем положениям, которые необходимы для грамотного подхода к работе с ферментами: подбору условий для определения активности фермента, определению начальной скорости ферментативной реакции, выбору субстрата, определению его насыщающей концентрации, оптимуму действия температуры и рН, влиянию кофакторов, активаторов и ингибиторов.
Наличие фермента в растворе или экстракте можно определить исходя из скорости катализируемой им реакции, о которой можно судить либо по накоплению продуктов реакции, либо по убыли субстрата.
В большинстве своем ферментативные реакции являются реакциями смешанного порядка. Типичная кривая хода ферментативной реакции (рис. 8.1) имеет следующий вид:
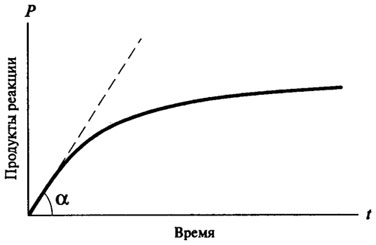
Рис. 8.1.Кривая хода ферментативной реакции во времени
Таким образом, ход ферментативной реакции во времени не может быть описан одним математическим уравнением, поскольку все ферментативные реакции в самом начале своего протекания (когда имеется избыток субстрата и образовалось мало продуктов реакции) являются
реакциями нулевого порядка, и только потом они приобретают характер реакции первого или второго порядка. Скорость реакции нулевого порядка со временем не меняется, зависимость количества образовавшегося продукта от времени остается прямо пропорциональной (см. рис. 8.2). Для реакций первого порядка скорость реакции в каждый данный момент времени пропорциональна имеющейся в наличии концентрации субстрата, а следовательно, наблюдается постоянное падение скорости реакции с течением времени (см. рис. 8.3).
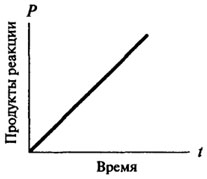
Рис. 8.2.Графическое изображение реакции нулевого порядка
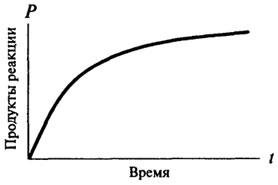
Рис. 8.3.Графическое изображение реакции первого порядка
Для того чтобы правильно определить потенциальные возможности данного фермента как катализатора, нужно учитывать скорость ферментативной реакции в тот момент времени, когда факторы, замедляющие скорость ферментативной реакции (нехватка субстрата, специфическое ингибирование продуктами реакции, частичная тепловая денатурация фермента и др.), не успевают проявить свое действие и наблюдается прямая пропорциональная зависимость между продуктами реакции и временем.
Такая скорость называется начальной скоростью ферментативной реакции и обозначается V0.
На практике V0 определяют графическим методом, для чего строят кривую хода ферментативной реакции во времени. Начальная скорость определяется как тангенс угла наклона касательной, проведенной из начала координат к кривой хода ферментативной реакции (см. рис. 8.1).
При работе с конкретным ферментом длительность реакции следует выбирать исходя из экспериментальных данных, по начальной скорости реакции.
В зависимости от задачи, которая стоит перед исследователями или технологами, теми, кто работает с ферментами, выбирается тот или иной подход в этой работе. Имеется в виду следующее.
1. Если необходимо выделить и охарактеризовать фермент из какого-либо биологического объекта, пищевого сырья, следует применить либо известные схемы выделения и очистки, или разработать оптимальную схему для данного фермента, варьируя и испытывая различные сочетания основных этапов очистки и выделения ферментов (белков): экстракцию, различные режимы осаждения, гель-хроматографию и другие методы, основанные на различиях в физико-химических характеристиках отдельных ферментов (см. также гл. 2). При этом на каждом этапе выделения и очистки следует характеризовать ферментный препарат по ферментативной активности и содержанию белка. В этом случае определение ферментативной активности (определение F0) проводят с использованием стандартного субстрата; выявляют оптимальные значения рН и температуры. И все дальнейшие исследования проводят при насыщающей концентрации субстрата, оптимуме температуры и рН. Изучение влияния специфических активаторов и ингибиторов позволяет в этом случае получить ценные сведения о строении активного центра и возможном механизме каталитического действия. Здесь необходимо подчеркнуть важность тщательного методического подхода при работе с ферментами. Не следует жалеть времени и усилий на выбор режима экстракции (продолжительность, температура, экстрагент, тип экстракции - исчерпывающая или нет), выбор методики определения активности, ее отработку и возможную модификацию для данного конкретного объекта исследования; кроме того, работа с ферментами различной степени очистки также имеет свои особенности, свою специфику: они обладают разной рН- и термостабильностью и, помимо этого, могут по-разному реагировать на воздействие различных факторов.
2. Если задача заключается в определении того, каким образом будет вести себя данный фермент (ферментный препарат) в конкретном режиме рассматриваемой пищевой технологии, необходимо проводить исследование ферментативного действия при условиях данного технологического процесса (концентрация субстрата, длительность, рН, температура, влажность), изучить влияние различных компонентов пищевого сырья и используемых добавок на активность фермента с целью определить возможность и способы влияния на ферментативный процесс в желаемом направлении.
Перейдем к рассмотрению факторов, влияющих на скорость ферментативных реакций.
Влияние концентрации субстрата на скорость ферментативной реакции.Концентрация субстрата является важнейшим фактором, определяющим скорость ферментативной реакции. Еще в 1902г. В. Анри при изучении реакции ферментативного гидролиза сахарозы предположил, что фермент р-фруктофуранозидаза взаимодействует со своим субстратом, затем это
соединение распадается, фермент остается в первоначальном виде, а субстрат сахароза оказывается расщепленной на глюкозу и фруктозу.
Это предложение было в дальнейшем развито Л. Михаэлисом и M. Ментен. В 1913 г. они постулировали следующие уравнения ферментативной реакции:
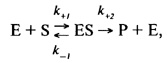
где k+1- константа скорости реакции образования комплекса ES, k-1 k+2 - константы скорости реакции распада комплекса ES в двух направлениях.
Тогда Ks- константа диссоциации комплекса ES равна отношению констант скоростей обратной и прямой реакции:
Исходя из закона действующих масс, можно записать следующее уравнение:
где [E0] - концентрация фермента в начале ферментативной реакции, [S] - концентрация субстрата, (ES] - концентрация комплекса "фермент-субстрат", [E0]-[ES] - концентрация фермента, не связанного в комплексе с субстратом.
В ходе ферментативной реакции в любой момент времени фермент существует в двух формах: свободной и связанной, т. е. в форме комплекса ES.
Скорость ферментативной реакции будет максимальной при такой концентрации субстрата, когда весь фермент перейдет в комплекс ES, т.е. когда все активные центры насыщены субстратом и дальнейшее увеличение концентрации субстрата не приведет к увеличению скорости реакции.
Преобразуя представленное выше уравнение, получим выражение, которое будет иметь следующий вид:
Это уравнение названо уравнением Михаэлиса-Ментен. Оно имеет огромное значение для выражения зависимости действия ферментов от концентрации субстрата. Однако оно содержит и ряд недостатков, в частности, при его выводе было сделано несколько допущений, например, не учитывалась вторая стадия ферментативной реакции - образование E и P.
В связи с этим был предложен рад усовершенствованных уравнений, с учетом влияния образовавшихся продуктов реакции. В настоящее время наиболее широко используют уравнение Холдейна-Бриггса. Оно имеет следующий вид:
В этом уравнении вместо K s - константы диссоциации комплекса ES, который присутствует в уравнении Михаэлиса-Ментен, стоит Km-константа Михаэлиса (в числителе которой находятся константы скоростей реакций, ведущих к распаду комплекса ES в двух направлениях):
Поскольку K s =
, то есть Km всегда больше Ks.
Для того, чтобы графическая зависимость, выражающая влияние концентрации субстрата на начальную скорость ферментативной реакции, из гиперболической преобразовалась в прямолинейную, что, очевидно, представляет большее удобство в экспериментальной практике, уравнение Холдейна-Бриггса было преобразовано Лайнуивером и Берком по методу двойных обратных величин.
Графически это выгладит так:
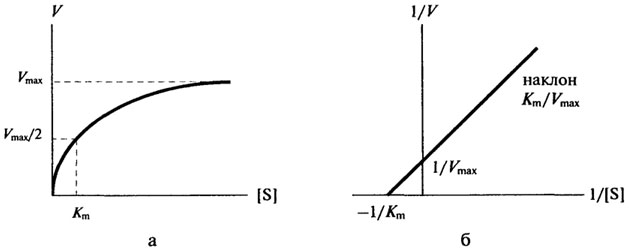
Рис. 8.4.Влияние концентрации субстрата на начальную скорость ферментативной реакции (а - по методу Михаэлиса-Ментен, б - по методу Лайнуивера-Берка)
Величина Km - это ключевой кинетический параметр; если [S] = K m, то V= Vmax/2, следовательно, константа Михаэлиса численно равна концентрации субстрата (в молях на литр), при которой скорость реакции равна половине максимальной.
Приблизительное значение Km можно получить простым графическим способом, как это показано на рис. 8.4 а; однако в этом способе достаточно велика погрешность в нахождении Vmax. Значительно удобнее пользоваться прямолинейной зависимостью при обработке данных по методу двойных обратных величин, рис. 8.4 б. В этом случае можно получить более точное значение Km.
Таблица 8.1.Значение констант Михаэлиса - К (мМ/л) для некоторых ферментов
| Фермент | Субстрат | К |
| Каталаза | H2 O 2 | 25,0 |
| Гексокиназа | АТФ | 0,4 |
| Глюкоза | 0,05 | |
| Фруктоза | 1,5 | |
| Химотрипсин | Глицил-тирозинил-глицин | 10,8 |
| N-бензол-тирозин-амид | 2,5 | |
| P - Галактозидаза | Лактоза | 4,0 |
Источником множества недоразумений как в прошлом, так и в настоящем, является некорректное использование термина "константа Михаэлиса" и двух символов Ks и Km для обозначения величин отнюдь неидентичных, несмотря на совершенно четкие рекомендации Комиссии по ферментам Международного Биохимического Союза. Первая величина -Ks- константа равновесия, выражаемая отношением Ks =
характеризует сродство фермента к субстрату (или, иначе, прочность комплекса ES), причем существует обратная пропорциональность между величиной Ks и сродством фермента к субстрату. Вторая величина -Km-соответствует концентрации субстрата, при которой V= Vmax/ 2. Часто свойство Ks ошибочно приписывают Km. Ha самом деле Km будет являться мерой сродства фермента к субстрату только в том единственном случае, когда величина k+2 будет настолько мала, что Km практически совпадет с Ks.
Многие ферменты катализируют реакции с участием двух субстратов. К так называемым бимолекулярным реакциям относятся реакции переноса химических группировок с одного соединения на другое, реакции синтеза, окислительно-восстановительные реакции.
Такие реакции могут протекать по двум различным механизмам. В реакциях первого типа, называемых реакциями единичного замещения, два субстрата А и В образуют с ферментом комплекс EAB, который затем распадается с образованием продуктов реакции С и Д. Второй тип двухсубстратных реакций протекает по механизму двойного замещения (механизм типа "пинг-понг"). В этих реакциях с активным центром фермента в каждый момент времени связан только один из двух субстратов.
При исследовании кинетики бимолекулярных реакций концентрацию одного из субстратов оставляют постоянной (В), а второго - изменяют (А). В этом случае в координатах 1/V от 1/[A] можно получить "кажущееся" значение К т. Истинное значение Vmax и К B mполучают при исследовании нескольких концентраций субстрата В. Точно так же поступают при определении K A m(когда концентрация А постоянна, а концентрация В варьируется). Кт по отношению к различным субстратам в одной и той же реакции могут быть различными - это хорошо видно из следующего примера.
Одним из характерных проявлений жизни является удивительная способность живых организмов кинетически регулировать химические реакции, подавляя стремление к достижению термодинамического равновесия. Ферментативная кинетика занимается исследованием закономерностей влияния химической природы реагирующих веществ (ферментов, субстратов) и условий их взаимодействия (концентрация, рН среды, температуры, присутствие активаторов или ингибиторов) на скорость ферментативной реакции. Главной целью изучения кинетики ферментативных реакций является получение информации, которая может способствовать выяснению молекулярного механизма действия фермента.
Общие принципы кинетики химических реакций применимы и к ферментативным реакциям. Известно, что любая химическая реакция характеризуется константой термодинамического равновесия. Она выражает состояние химического равновесия, достигаемого системой, и обозначается Кр. Так, для реакции:
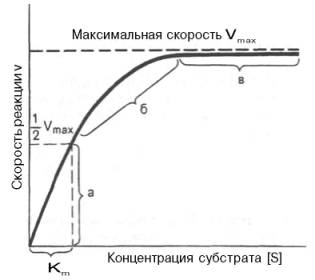
Рис. 4.12. Теоретический график зависимости скорости ферментативной реакции от концентрации субстрата при постоянной концентрации фермента.
а - реакция первого порядка (при [ S ]
Таким образом, константа равновесия равна отношению констант скоростей прямой и обратной реакций. Величину, обратную константе равновесия, принято называть субстратной константой, или, в случае ферментативной реакции, константой диссоциации фермент–субстратного комплекса, и обозначать символом KS. Так, в реакции
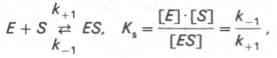
т.е. KSравна отношению произведения концентрации фермента и субстрата к концентрации фермент-субстратного комплекса или отношению констант скоростей обратной и прямой реакций. Следует отметить, что константа KSзависит от химической природы субстрата и фермента и определяет степень их сродства. Чем ниже значение KS, тем выше сродство фермента к субстрату.
При изучении кинетики ферментативных реакций следует учитывать одну важную особенность этих реакций (не свойственную обычным химическим реакциям), связанную с явлением насыщения фермента субстратом. При низкой концентрации субстрата зависимость скорости реакции от концентрации субстрата (рис. 4.12) является почти линейной и подчиняется кинетике первого порядка. Это означает, что скорость реакции S —> Р прямо пропорциональна концентрации субстрата S и в любой момент времени t определяется следующим кинетическим уравнением:
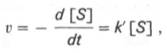
где [S] – молярная концентрация субстрата S; –d[S]/dt – скорость убыли субстрата; k' – константа скорости реакции, которая в данном случае имеет размерность, обратную единице времени (мин –1 или с –1 ).
При высокой концентрации субстрата скорость реакции максимальна, становится постоянной и не зависящей от концентрации субстрата [ S ] . В этом случае реакция подчиняется кинетике нулевого порядка v = k" (при полном насыщении фермента субстратом) и целиком определяется концентрацией фермента. Различают, кроме того, реакции второго порядка, скорость которых пропорциональна произведению концентраций двух реагирующих веществ. В определенных условиях при нарушении пропорциональности говорят иногда о реакциях смешанного порядка (см. рис. 4.12).
Изучая явление насыщения, Л. Михаэлис и М. Ментен разработали общую теорию ферментативной кинетики. Они исходили из предположения, что ферментативный процесс протекает в виде следующей химической реакции:
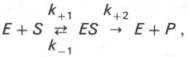
т.е. фермент Е вступает во взаимодействие с субстратом S с образованием промежуточного комплекса ES, который далее распадается на свободный фермент и продукт реакции Р. Математическая обработка на основе закона действующих масс дала возможность вывести уравнение, названное в честь авторов уравнением Михаэлиса–Ментен, выражающее количественное соотношение между концентрацией субстрата и скоростью ферментативной реакции:
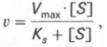
Из уравнения Михаэлиса–Ментен следует, что при высокой концентрации субстрата и низком значении KSскорость реакции является максимальной, т.е. v = Vmax(реакция нулевого порядка, см. рис. 4.12). При низкой концентрации субстрата, напротив, скорость реакции оказывается пропорциональной концентрации субстрата в каждый данный момент (реакция первого порядка).
Следует указать, что уравнение Михаэлиса–Ментен в его классическом виде не учитывает влияние на скорость ферментативного процесса продуктов реакции, например в реакции
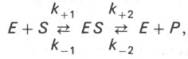
и носит несколько ограниченный характер. Поэтому были предприняты попытки усовершенствовать его. Так, было предложено уравнение Бриггса-Холдейна:
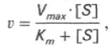
где Кm представляет собой константу Михаэлиса, являющуюся экспериментально определяемой величиной. Она может быть представлена следующим уравнением:
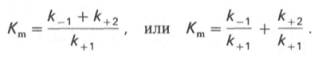
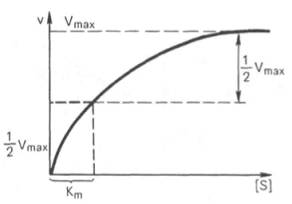
Рис. 4.13. Кривая уравнения Михаэли-са-Ментен: гиперболическая зависимость начальных скоростей катализируемой ферментом реакции от концентрации субстрата.
В числителе представлены константы скоростей распада комплекса ES в двух направлениях (в сторону исходных Е и S и в сторону конечных продуктов реакции Е и Р). Отношение k–1/ k+1представляет собой константу диссоциации ферментсубстратного комплекса KS, тогда:
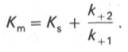
Отсюда вытекает важное следствие: константа Михаэлиса всегда больше константы диссоциации фермент-субстратного комплекса KSна величину
Для определения численного значения Кm обычно находят ту концентрацию субстрата, при которой скорость ферментативной реакции v составляет половину от максимальной Vmax, т.е. если v = 1 /2 Vmaх. Подставляя значение v в уравнение Бриггса–Холдейна, получаем:
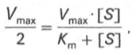
разделив обе части уравнения на Vmах, получим
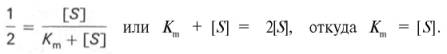
Таким образом, константа Михаэлиса численно равна концентрации субстрата (моль/л), при которой скорость данной ферментативной реакции составляет половину от максимальной.
Определение величины Кm имеет важное значение при выяснении механизма действия эффекторов на активность ферментов и т.д. Константу Михаэлиса можно вычислить по графику (рис. 4.13). Отрезок на абсциссе, соответствующий скорости, равной половине максимальной, будет представлять собой Кm.
Пользоваться графиком, построенным в прямых координатах зависимости начальной скорости реакции v0 от начальной концентрации субстрата [S0], неудобно, поскольку максимальная скорость Vmaxявляется в данном случае асимптотической величиной и определяется недостаточно точно.
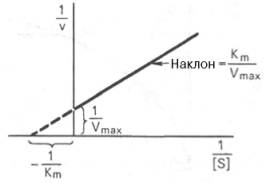
Рис. 4.14. График Лайнуивера-Бэрка.
Для более удобного графического представления экспериментальных данных Г. Лайнуивер и Д. Бэрк преобразовали уравнение Бриггса–Хол-дейна по методу двойных обратных величин исходя из того принципа, что если существует равенство между двумя какими-либо величинами, то и обратные величины также будут равны. В частности, если
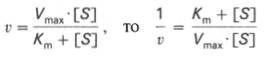
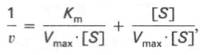
то после преобразования получаем уравнение:
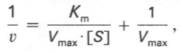
которое получило название уравнения Лайнуивера–Бэрка. Это уравнение прямой линии: у = ах + b. Если теперь в соответствии с этим уравнением построить график в координатах 1/v (y) от l/[S] (x), то получим прямую линию (рис. 4.14), тангенс угла наклона который будет равен величине Km/Vmax; отрезок, отсекаемый прямой от оси ординат, представляет собой l/Vmax(обратная величина максимальной скорости). Если продолжить прямую линию за ось ординат, тогда на абсциссе отсекается отрезок, соответствующий обратной величине константы Михаэлиса – 1/Кm (см. рис. 4.14). Таким образом, величину Кm можно вычислить из данных наклона прямой и длины отрезка, отсекаемого от оси ординат, или из длины отрезка, отсекаемого от оси абсцисс в области отрицательных значений.
Следует подчеркнуть, что значения Vmax, как и величину Кm, более точно, чем по графику, построенному в прямых координатах, можно определить по графику, построенному по методу двойных обратных величин. Поэтому данный метод нашел широкое применение в современной энзимологии. Предложены также аналогичные графические способы определения Кm и Vmaxв координатах зависимости v от v/[S] и [S]/v от [S].
Следует отметить некоторые ограничения применения уравнения Ми-хаэлиса–Ментен, обусловленные множественными формами ферментов и аллостерической природой фермента. В этом случае график зависимости начальной скорости реакции от концентрации субстрата (кинетическая
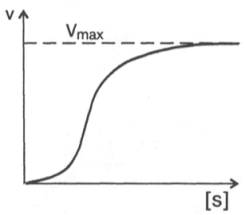
Рис. 4.15. Сигмоидная кинетическая кривая насыщения субстратом.
кривая) имеет не гиперболическую форму, а сигмоидный характер (рис. 4.15) наподобие кривой насыщения гемоглобина кислородом. Это означает, что связывание одной молекулы субстрата в одном каталитическом центре повышает связывание субстрата с другим центром, т.е. имеет место кооперативное взаимодействие, как и в случае присоединения кислорода к 4 субъединицам гемоглобина. Для оценки концентрации субстрата, при которой скорость реакции составляет половину максимальной, в условиях сигмоидного характера кинетической кривой обычно применяют преобразованное уравнение Хилла:
Характер зависимости ферментативной реакции от рН определяется тем, что этот показатель оказывает влияние на:
a) ионизацию аминокислотных остатков, участвующих в катализе,
b) ионизацию субстрата,
c) конформацию фермента и его активного центра.
Ингибирование ферментов
Скорость ферментативной реакции может быть снижена действием ряда химических веществ, называемых ингибиторами. Некоторые ингибиторы являются для человека ядами, например, цианиды, другие – используются в качестве лекарственных препаратов.
Ингибиторы можно разделить на два основных типа: необратимые и обратимые. Необратимые ингибиторы (I) связываются с ферментом с образованием комплекса, диссоциация которого с восстановлением активности фермента невозможна:
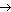
E + I EI.
Примером необратимого ингибитора является диизопропилфторфосфат (ДФФ). ДФФ ингибирует фермент ацетилхолинэстеразу, играющего важную роль в передаче нервного импульса. Этот ингибитор взаимодействует с серином активного центра фермента, блокируя тем самым активность последнего. Вследствие этого нарушается способность отростков нервных клеток нейронов проводить нервный импульс. ДФФ является одним из первых веществ нервно-паралитического действия. На его основе создан ряд относительно нетоксичных для человека и животных инсектицидов - веществ, ядовитых для насекомых.
Обратимые ингибиторы, в отличие от необратимых, при определенных условиях могут быть легко отделены от фермента. Активность последнего при этом восстанавливается:
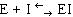
.
Среди обратимых ингибиторов выделяют конкурентные и неконкурентные ингибиторы.
Конкурентный ингибитор, являясь структурным аналогом субстрата, взаимодействует с активным центром фермента и таким образом перекрывает доступ субстрата к ферменту. При этом ингибитор не подвергается химическим превращениям и связывается с ферментом обратимо. После диссоциации комплекса EI фермент может связаться либо с субстратом и преобразовать его, либо с ингибитором (рис. 34.). Поскольку и субстрат и ингибитор конкурируют за место в активном центре, такое ингибирование называется конкурентным.
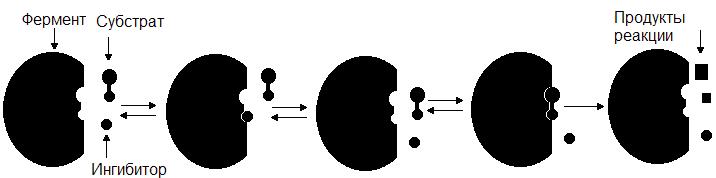
Рис. 34. Механизм действия конкурентного ингибитора.
Конкурентные ингибиторы используются в медицине. Для борьбы с инфекционными болезнями ранее широко применялись сульфаниламидные препараты. Они близки по своей структуре к пара-аминобензойной кислоте (ПАБК), необходимому фактору роста многих патогенных бактерий. ПАБК является предшественником фолиевой кислоты, которая служит кофактором ряда ферментов. Сульфаниламидные препараты выступают в качестве конкурентного ингибитора ферментов синтеза фолиевой кислоты из ПАБК и тем самым подавляют рост и размножение патогенных бактерий.
Неконкурентные ингибиторы по структуре не сходны с субстратом и при образовании EI взаимодействуют не с активным центром, а с другим участком фермента. Взаимодействие ингибитора с ферментом приводит к изменению структуры последнего. Образование EI-комплекса является обратимым, поэтому после его распада фермент вновь способен атаковать субстрат (рис. 35).
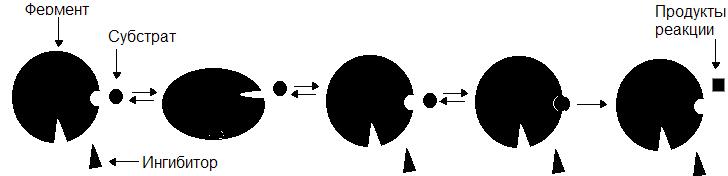
Рис. 35. Механизм действия неконкурентного ингибитора
В качестве неконкурентного ингибитора может выступать цианид CN - . Он связывается с ионами металлов, входящими в состав простетических групп и подавляет активность этих ферментов. Отравления цианидами крайне опасны. Они могут привести к летальному исходу.
Аллостерические ферменты
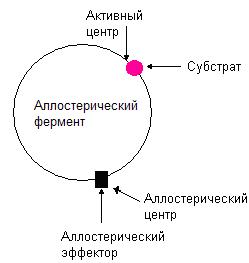
Рис. 36. Структура аллостерического фермента.
Регуляция мультиферментных систем
Некоторые ферменты действуют согласованно, объединяясь в мультиферментные системы, в которых каждый фермент катализирует определенную стадию метаболитического пути:
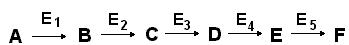
В мультиферментной системе есть фермент, который определяет скорость всей последовательности реакций. Этот фермент, как правило, бывает аллостерическим и находится в начале матаболитического пути. Он способен, получая различные сигналы, как повышать, так и понижать скорость катализируемой реакции, тем самым регулируя скорость всего процесса.
Ферментативная кинетика изучает влияние различных факторов (концентрация S и E, рН, температура, давление, ингибиторы и активаторы) на скорость ферментативных реакций. Главной целью изучения кинетики ферментативных реакций является получение информации, позволяющей глубже понять механизм действия ферментов.
Кинетическая кривая позволяет определить начальную скорость реакции V0.

Кривая субстратного насыщения.

График зависимости V от концентрации субстрата при фиксированной концентрации фермента представляет собой гиперболу. Вначале скорость реакции прямо пропорциональна концентрации субстрата [S] (кинетика первого порядка). Однако при увеличении [S] скорость постепенно достигает максимального значения VMAX. Это означает, что все связывающие участки фермента заняты (насыщены). Скорость реакции на этом участке не зависит от концентрации субстрата (кинетика нулевого порядка). Такую кривую называют кривой субстратного насыщения.
Зависимость скорости реакции от концентрации фермента.

При постоянной концентрации субстрата существует прямо пропорциональная зависимость между скоростью реакции и концентрацией фермента [E] в реакционной смеси. Другими словами, для данной концентрации субстрата скорость реакции возрастает в 2 раза при двукратном увеличении концентрации фермента.
Зависимость скорости реакции от температуры.

Ферменты – вещества белковой природы, проявляют максимальную активность в ограниченном температурном режиме. При температурах не выше 40-50С скорость реакции увеличивается согласно теории химической кинетики. При более высоких температурах тепловая денатурация фермента приводит к полному прекращению ферментативной реакции. Термолабильность ферментов отличает ферменты от неорганических катализаторов.
Зависимость скорости реакции от рН.

Оптимум рН действия большинства ферментов лежит в пределах физиологических значений 6,0-8,0. Пепсин активен при рН 1,5-2,0, что соответствует кислотности желудочного сока. Аргиназа, специфичный фермент печени, активен при 10,0. Влияние рН среды на скорость ферментативной реакции связывают с состоянием и степенью ионизации ионогенных групп в молекуле фермента и субстрата. Этот фактор определяет конформацию белка, состояние активного центра и субстрата, формирование фермент-субстратного комплекса, собственно процесс катализа.
Математическое описание кривой субстратного насыщения, константа Михаэлиса.

Уравнение, описывающее кривую субстратного насыщения, было предложено Михаэлисом и Ментон и носит их имена (уравнение Михаэлиса-Ментен):
V = (VMAX*[S])/(Km+[S]), где Km – константа Михаэлиса. Легко рассчитать, что при V = VMAX/2 Km = [S], т.е. Km – это концентрация субстрата, при которой скорость реакции составляет ½ VMAX.
С целью упрощения определения величины VMAX и Km уравнение Михаэлиса-Ментен можно пересчитать.
1/V = Km/(VMAX*[S]) + 1/VMAX,

1/V = Km/VMAX*1/[S] + 1/VMAX уравнение Лайнуивера-Берка. Уравнение, описывающее график Лайнуивера-Берка – это уравнение прямой линии (y = mx + c), где 1/VMAX – это отрезок, отсекаемый прямой на оси ординат; Km/VMAX - тангенс угла наклона прямой; пересечение прямой с осью абсцисс дает величину 1/Km. График Лайнуивера-Бэрка позволяет определить Km по относительно небольшому числу точек. Этот график также используют при оценке действия ингибиторов, о чем будет сказано ниже.
Значение Km изменяются в широких пределах: от 10 -6 моль/л для очень активных ферментов, до 10 -2 – для малоактивных ферментов.
Оценки Km имеют практическую ценность. При концентрациях субстрата в 100 раз превышающих Km, фермент будет работать практически с максимальной скоростью, поэтому максимальная скорость VMAX будет отражать количество присутствующего активного фермента. Это обстоятельство используют для оценки содержания фермента в препарате. Кроме того, Km является характеристикой фермента, что используется для диагностики энзимопатий.
Ингибирование активности ферментов.
Чрезвычайно характеристикой и важной особенностью ферментов является их инактивация под влиянием определенных ингибиторов.
Ингибиторы – это вещества, вызывающие частичное или полное торможение реакций, катализируемых ферментами.
Ингибирование ферментативной активности может быть необратимым или обратимым, конкурентным или неконкрентным.
Необратимое ингибирование – это стойкая инактивация фермента, возникающая в результате ковалентного связывания молекулы ингибитора в активном центре или в другом особом центре, изменяющим конформацию фермента. Диссоциация столь устойчивых комплексов с регенерацией свободного фермента практически исключена. Для преодоления последствий такого ингибирования организм должен синтезировать новые молекулы фермента.
Обратимое ингибирование – характеризуется равновесным комплексообразованием ингибитора с ферментом за счет нековалентных связей, вследствие чего такие комплексы способны к диссоциации с восстановлением активности фермента.
Классификация ингибиторов на конкурентные и неконкурентные основана на том, ослабляется (конкурентное ингибирование) или не ослабляется (неконкурентное ингибирование) их ингибирующие действие при повышении концентрации субстрата.
Конкурентные ингибиторы – это, как правило, соединения, структура которых сходна со структурой субстрата. Это позволяет им связываться в том же активном центре, что и субстраты, препятствуя взаимодействию фермента с субстратом уже на стадии связывания. После связывания ингибитор может быть превращен в некий продукт или остается в активном центре, пока не произойдет диссоциация.
Обратимое конкурентное ингибирование можно представить в виде схемы:

E↔ E-I → E + P1
+S ( неакт )
Степень ингибирования фермента определяется соотношением концентраций субстрата и фермента.
Классическим примером подобного типа ингибирования является торможение активности сукцинатдегидрогеназы (СДГ) малатом, который вытесняет сукцинат из субстратного участка и препятствует его превращению в фумарат:

Ковалентное связывание ингибитора в активном центре приводит к инактивации фермента (необратимое ингибирование). Примером необратимого конкурентного ингибирования может служить инактивация триозофосфатизомеразы 3-хлорацетолфосфатом. Этот ингибитор является структурным аналогом субстрата – диоксиацетонфосфата и необратимо присоединяется к остатку глутаминовой кислоты в активном центре:

Некоторые ингибиторы действуют менее избирательно, взаимодействуя с определенной функциональной группой в составе активного центра разных ферментов. Так, связывание йодацетата или его амида с SH-группой аминокислоты цистеина, находящийся в активном центре фермента и принемающей участие в катализе, приводит к полной утрате активности фермента:
Поэтому эти ингибиторы инактивируют все ферменты, которые имеют SH-группы, участвующие в катализе.
Необратимое ингибирование гидролаз при действии нервно-паралитических газов (зарин, зоман) обусловлено их ковалентным связыванием с остатком серина в активном центре.
Метод конкурентного ингибирования нашел широкое применение в медицинской практике. Сульфаниламидные препараты – антагонисты п-аминобензойной кислоты, могут служить примером метаболизируемых конкурентных ингибиторов. Они связываются с дигидроптератсинтетазой – бактериальным ферментом, осуществляющим превращение п-аминобензоата в фолиевую кислоту, необходимую для роста бактерий. Бактерия погибает в результате того, что связавшийся сульфаниламид превращается в другое соединение и фолиевая кислота не образуется.
Неконкурентные ингибиторы обычно связываются с молекулой фермента в участке, отличном от места связывания субстрата, и субстрат непосредственно не конкурирует с ингибитором. Поскольку ингибитор и субстрат связываются с разными центрами возможно образование как комплекса E-I, так и комплекса S-E-I. Комплекс S-E-I тоже распадается с образованием продукта, однако с меньшей скоростью, чем E-S, поэтому реакция будет замедляться, но не остановится. Таким образом, могут протекать следующие параллельные реакции:
E↔ E-I ↔ S-E-I → E-I + P
Обратимое неконкурентное ингибирование встречается сравнительно редко.
Неконкурентные ингибиторы называют аллостерическими в отличие от конкурентных (изостерических).
Обратимое ингибирование может быть количественно изучено на основе уравнения Михаэлиса-Ментен.
При конкурентном ингибировании VMAX остается постоянной, а Km возрастает.


При неконкурентном ингибировании снижается VMAX при неизменном Km.


Если продукт реакции ингибирует фермент, катализирующий его образование, такой способ ингибирования называется ретроингибированием или ингибированием по принципу обратной связи. Например, глюкоза тормозит глюкозо-6-фосфатазу, которая катализирует гидролиз глюкозо-6-фосфата.
Биологическое значение такого ингибирования – регуляция определенных метаболических путей (см. следующее занятие).
1. Изучить денатурацию белков под действием растворов минеральных и органических кислот и при нагревании.
2. Обнаружить кофермент НАД в дрожжах.
3. Определить амилазную активность в моче (сыворотке крови).
10. ХАРАКТЕР И ОБЪЕМ ВОЗМОЖНОЙ УЧЕБНО-ИССЛЕДОВАТЕЛЬСКОЙ РАБОТЫ ПО ТЕМЕ
(Указать конкретно характер и форму УИРС: подготовка реферативных выступлений, проведение самостоятельных исследований, имитационная игра, оформление истории болезни с использованием монографической литературы и др. формы)
11. ЛИТЕРАТУРА ДЛЯ ПОДГОТОВКИ К ЗАНЯТИЮ ПРЕПОДАВАТЕЛЯМ:
Тут вы можете оставить комментарий к выбранному абзацу или сообщить об ошибке.
Читайте также: