
Болезнь тея сакса реферат
Обновлено: 08.07.2024
Первая (наиболее распространённая) начинает развиваться почти сразу после рождения ребёнка, и длительность жизни малыша в таком случае составляет 4-5 лет.
Болезнь Тея-Сакса с поздним началом проявляется между периодом полового созревания и возрастом 23-26 лет. Продолжительность жизни больного зависит в первую очередь от тяжести симптомов заболевания. Такие пациенты могут прожить тот же промежуток времени, что и здоровый человек.
Что может вызывать эту болезнь?
Болезнь Тея-Сакса может развиваться, если родители передали ребёнку поражённый (мутировавший) ген .
Они входят в состав клеток организма и содержат генетическую информацию или дезоксирибонуклеиновую кислоту (ДНК), которая отвечает за физические характеристики человека. Гены (по отдельности или в совокупности) определяют какие генетические черты ребёнок унаследует от родителей, на пример, группу крови, цвет волос, глаз, а также вероятность развития того или иного заболевания. Определённое количество генов составляют хромосому. Поражения как гена, так и хромосомы, вызывают изменения в процессах и функциях организма. Эти дефекты могут практически не проявляться, однако иногда они становятся причиной развития таких серьёзных заболеваний как гемофилия или синдром Дауна. Такие проблемы со здоровьем как болезнь Стилла (форма ревматоидного артрита у детей) или депрессия также могут быть следствием отклонений на генетическом уровне. Мутировавший ген переходит от родителей к детям. Болезни, передающиеся по наследству, обычно имеют генетический характер. Человек может родиться с генетическим фоном, который будет влиять на повышенную восприимчивость или риск проявления определённого заболевания.
Когда малыш унаследует искажённый ген от обоих родителей, он заболевает.
Если происходит передача поражённого гена от одного из родителей – ребёнок становится носителем дефекта . Это означает, что у него будет присутствовать данный ген в организме, однако сам он не заболеет.
Это человек, который может передать своим детям наследственную (генетическую) болезнь, хотя сам и не является поражённым недугом. Кроме этого, существует также риск передачи статуса носителя заболевания. Некоторые недуги вызваны именно отклонениями на генном и хромосомном уровнях. Каждый человек унаследует 23 хромосомы от каждого из родителей. Таким образом, у него получается 23 пары хромосом в организме. Если одна или обе хромосомы из пары поражены, возникает генетическое заболевание. В большинстве случаев, у человека с вышеуказанным дефектом обе хромосомы в паре являются повреждёнными. Такое расстройство называется болезнью с аутосомно-рецессивным типом наследования. Определённые генетические заболевания возникают из-за поражения X и Y хромосом, которые отвечают за определение пола. Если хотя бы одна хромосома из пары с отклонением, человек может быть носителем заболевания.
Мутировавший ген препятствует выработке в организме такого фермента как гексозаминидаза А, который отвечает за расщепление ганглиозидов (сложных природных липидов) в клетках. В случае их накопления происходит блокирование работы мозга и повреждение нервных клеток, что и вызывает болезнь Тея-Сакса.
Ферменты Это белок, который вырабатывается для ускорения протекания определённой химической реакции. В нашем организме существует много ферментов для обеспечения различных процессов жизнедеятельности, таких как пищеварение и свёртывание крови. Некоторые генетические заболевания напрямую связаны с выработкой определённых ферментов. Чтобы установить диагноз (на пример, проблемы с почками) врач измеряет количество необходимого фермента в крови пациента.
При болезни Тея-Сакса с поздним началом в организме производится пониженное количество вышеуказанного фермента. Люди с таким заболеванием унаследуют ген гексозаминидаза А с поздним развитием от обоих родителей или же по одному гену с поздним развитием и неспособностью к активности. Риск появления мутировавшего гена возрастает у людей с корнями, которые уходят к евреям-ашкенази, так как 1 из 30 представителей этой национальности является носителем данного заболевания. Кроме того, к развитию болезни Тея-Сакса также предрасположены люди французско-канадского происхождения, проживающие в восточной части долины реки Святого Лоренса провинции Квебек и каджуны (франкоязычные жители штата Луизиана).
Болезнь Тея-Сакса – симптомы
В 3-6 месяцев малыш начинает слабо зрительно реагировать на действия вокруг него, и ему сложно сфокусироваться на предмете. Врач может обнаружить красные точки на сетчатке глаза ребёнка.
В 6-10 месяцев у ребёнка происходит спад активности. Ему становится сложно сидеть и переворачиваться. Кроме этого, возникают проблемы со слухом и зрением.
После 10 месяцев болезнь активно прогрессирует. У ребёнка возможны приступы , потеря зрения и атрофия мышц.
Это внезапные вспышки аномальной электрической активности мозга, которые влияют на функционирование мышечной системы, контроль двигательного аппарата, искажают речь, зрительные образы и психологическое восприятие действительности. Степень поражения у каждого индивидуальная. Это также зависит от типа, частоты и тяжести припадков. В некоторых случаях человек падает на пол и начинает биться в конвульсиях, при которых мышцы тела сильно сокращаются, и возникает непроизвольное подёргивание конечностей. Другие входят в состояние транса, у них остаются подвижными лишь несколько мышц. Иногда они ощущают запахи или видят образы, нехарактерные для здорового человека. Часто припадок является симптомом других проблем со здоровьем, таких как возникновение высокой температуры тела (особенно у детей), нарушение мозгового кровообращения, инфекция, низкий уровень глюкозы в крови (гипогликемия), пониженное давление или опухоль мозга.
В ходе развития у малыша болезни Тея-Сакса с поздним началом такие симптомы как неуклюжесть и перепады настроения могут не восприниматься серьёзно. К более поздним признакам заболевания относятся судорожное подёргивание и слабость мышц, невнятная речь и проблемы с мыслительными процессами. Характер осложнений зависит от того, какое количество гексозаминидазы А вырабатывается в организме.
Болезнь Тея-Сакса – система распознавания заболевания
Если у лечащего врача есть подозрение, что ребёнок поражён болезнью Тея-Сакса, он прежде всего проведёт обследование пациента и возьмёт анализ крови, чтобы проверить уровень гексозаминидазы А в организме. Генетический тест необходим для подтверждения диагноза.
В чём заключается лечение?
По мере развития заболевания ребёнку может понадобиться более тщательный уход. Подбадривайте его и внушайте, что нужно бороться, подарите свою любовь и ласку. Возможно, у вас возникнет необходимость в дополнительной помощи по уходу за малышом. Проконсультируйтесь с лечащим врачом о том, куда можно обратиться по этому вопросу.
Болезнь Тея-Сакса - зачем нужно проходить обследование на наличие заболевания?
Носители данного порока могут передать детям заболевание даже если они сами не больны. Если вы и партнёр являетесь носителями болезни Тея-Сакса, риск развития аналогичного дефекта у ваших детей повышается до 1 из 4 случаев (25%). Когда вы планируете заводить ребёнка, Американская корпорация акушёров и гинекологов рекомендует сделать следующее:
Оба партнёра должны пройти скрининг тесты, если они относятся к таким категориям, как евреи-ашкенази, каджуны (франкоязычные жители штата Луизиана), имеют французско-канадское происхождение, либо же они имеют семейный анамнез по данной болезни. В случае если обследование покажет, что вы позитивны, последующие решения поможет принять прохождение генетического консультирования .
Если хотя бы один из партнёров соответствует вышеуказанным категориям, он также должен пройти процедуру тестирования. Один положительный результата на носителя заболевания предполагает также обследование другого партнёра.
Из семейного анамнеза врач может узнать, есть ли у пациента кровные родственники с аналогичным заболеванием. Это поможет специалисту установить степень риска передачи порока по наследству.
Насколько у пациента близкое родство с членом семьи, у которого было отмечено соответствующее заболевание.
Генетическое консультирование – это пояснения медицинского работника (консультанта по генетическим вопросам или генетика), специализация которого, помогать людям разобраться в опасности передачи генетических заболеваний, а также объяснять возможность унаследования ребёнком такого вида болезни (серповидно-клеточная болезнь, кистозный фиброз, гемофилия).
Объяснение родителям (будущим) как определённое заболевание может наследоваться и передаваться от них к ребёнку.
Рассмотрение, на основании полученных результатов, возможности рождения у пары ребёнка с генетическим заболеванием.
Помощь в осознании риска развития такого генетического заболевания как, на пример, болезнь Хантингтона. Консультанты по генетическим вопросам также помогают человеку с лечением генетического заболевания, если есть такая потребность.
Помощь, как отдельным пациентам, так и семейным парам в принятии решения в пользу проверки на генетическое заболевание.
Болезнь Тея-Сакса – это генетическое заболевание, характеризующееся недостаточностью фермента гексозаминидазы А, скоплением липоидных макромолекул в нейронах, нарушением функций головного и спинного мозга. Проявляется деградацией физических навыков и психических функций: распадом глотательного рефлекса, речи и произвольных движений, утратой слуха и зрения, снижением интеллекта. Развиваются судорожные приступы, атрофия мышц, паралич, деменция. Специфические методы диагностики – офтальмоскопия глазного дна, исследование количества гексозаминидазы в крови и нейронах. Лечение паллиативное, нацелено на облегчение симптомов.
МКБ-10
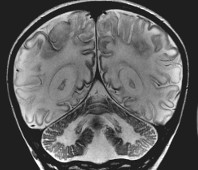
Общие сведения
Синонимы болезни Тея-Сакса (БТС) – ганглиозидоз GM2, идиотия Тея-Сакса, ранняя детская амавротическая идиотия. Является одним из вариантов лизосомных болезней накопления. Названа по фамилиям двух врачей – невропатолога из США Бернарда Сакса и офтальмолога из Великобритании Уоррена Тея. В 80-е годы XIX века они впервые опубликовали независимые описания данной патологии. Ее распространенность крайне низка, в общей популяции средняя частота носителей рецессивного измененного гена составляет около 0,3%. Эпидемиологические показатели наиболее высоки в группе евреев ашкенази (носительство мутации определяется у 3%), а также у франкоканадцев и каджунов. Пик заболеваемости приходится на возраст от полугода до 2 лет, реже симптомы дебютируют у подростков и взрослых.
Причины
К началу 2000-х годов выявлено более 100 различных мутаций в генах HEXA: вставки в пары оснований, делеции пар оснований, сплайс-сайт мутации, точечные мутации и другие варианты изменений структуры гена. Каждая из этих мутаций определенным образом влияет на строение фермента и подавляет его активность. Разнообразие мутаций пары генов – компаундная гетерозиготность – определяет наличие нескольких форм болезни. Одинаково мутировавшие гены в аллели вызывают полную инактивацию катаболизма ганглиозидов. Наследование разных (не одинаковых) мутаций в паре генов чаще проявляется уменьшением активности фермента, а не полной инактивацией.
Патогенез
Основой развития болезни является отсутствие или недостаточная активность гексозаминидазы A – лизосомального фермента, катализирующего биодеградацию макромолекул ганглиозидов, олигосахаридов, гликозаминогликанов и гликолипидов. Ганглиозиды – тип жирных кислот, липидные компоненты мембран нейронов и глиальных клеток. Они обеспечивают активность нервных клеток: влияют на скорость и интенсивность нейропередачи, проведение нервных импульсов, распределение и хранение информации, формирование памяти. В норме ганглиозиды производятся, выполняют свои функции и быстро расщепляются в процессе многоступенчатой реакции с участием ферментов. Для гидролиза этих сложных липидов необходимы три компонента: альфа- и бета-субъединицы гексозаминидазы А, белок-активатор GM2A. При недостаточности альфа-гексозаминидазы А процесс биодеградации замедляется или становится полностью невозможным. Ганглиозиды накапливаются в лизосомах клеток головного, спинного мозга, приводят к их дисфункции и гибели.
Классификация
В зависимости от особенностей генетического дефекта, сохранности функций гексозаминидазы заболевание имеет быстропрогрессирующее или медленно прогрессирующее течение, проявляется в раннем детстве, подростничестве или во взрослом возрасте. Согласно этим характеристикам выделено 3 формы болезни:
- Острая инфантильная. Наиболее распространенная. Симптомы возникают через несколько месяцев после рождения. Течение прогрессирующее. Быстро ухудшаются двигательные навыки, развивается слепота, глухота, паралич. Смерть наступает в течение 2-3 лет.
- Поздняя ювенильная. Встречается крайне редко. Первые проявления обнаруживаются в промежутке от 2 до 10 лет. Постепенно распадаются приобретенные сложные навыки – ходьба, речь, письмо. Средняя продолжительность жизни больных – 15 лет.
- Хроническая взрослая. В научной литературе описаны единичные случаи данной формы, которая начинается в 25-30 лет. Характеризуется нарушением речи, произвольных двигательных актов, расстройствами психики (психозы). Прогноз летальности неизвестен.
Симптомы болезни Тея-Сакса
Клиническая картина болезни отражает процессы поражения ЦНС. При инфантильной форме первые симптомы становятся заметными к 3-5 месяцам, до этого развитие соответствует норме: ребенок держит голову, переворачивается на живот и обратно, агукает, улыбается при виде взрослого, устанавливает визуальный контакт. К 6 месяцам снижается заинтересованность окружающим миром. Малыш подолгу смотрит в сторону, апатичен, малоподвижен, чувствителен к громким звукам, яркому свету. Он перестает узнавать близких людей, с трудом фокусирует взгляд на любимых игрушках.
Ювенильная форма дебютирует менее явными симптомами. На начальном этапе несколько усиливается эмоциональная неустойчивость, при выполнении сложных двигательных комплексов – беге, ходьбе, быстром письме – появляется едва заметная дискоординация движений. Через некоторое время неуклюжесть и неловкость нарастают, замечаются окружающими и самим ребенком. К подростковому возрасту походка становится шаткой, неустойчивой. Формируются гиперкинезы – внезапные непроизвольные движения различных групп мышц. Нарушения координации не позволяют продолжать школьное обучение. Параллельно появляются расстройства речи сложного мозжечково-дизартрического характера: теряется плавность и ритмичность, произношение становится медленным, смазанным, невнятным. Поздние стадии болезни характеризуются частыми эпилептическими приступами, стойким снижением интеллектуальных функций (деменцией), утратой произвольности движений, параличом.
Хроническая форма заболевания имеет менее выраженные симптомы, течение сравнительно легкое. У пациентов наблюдаются перепады настроения, неуклюжесть, ухудшается произношение. На протяжении нескольких лет снижаются интеллектуальные функции: утрачивается способность к абстрактному мышлению, сравнению и анализу явлений и предметов, нарастает забывчивость и рассеянность. Спустя несколько лет после начала болезни развиваются психические расстройства: больные неадекватны в поведении, аффективно возбудимы, склонны к состояниям ажитации и глубокой депрессии, подвержены психозам с галлюцинациями и бредом. При длительном течении формируется органическая деменция.
Осложнения
В число симптомов БТС входят эпилептические приступы, представляющие собой результат внезапных вспышек аномальной биоэлектрической активности мозга. При их высокой частоте физическая и психическая деградация происходит быстрее. Во время приступа больные падают, бьются в конвульсиях, что сопровождается высоким риском удушья (западение корня языка), получения смертельных травм. Основным осложнением острой инфантильной формы заболевания являются инфекции: у детей снижены функции иммунной системы, поражение органов дыхания носит рецидивирующий характер, протекает крайне тяжело. Распространенная причина смерти – пневмония.
Диагностика
Обследование проводится детским неврологом, офтальмологом, генетиком, психиатром. Процесс постановки диагноза начинается со сбора клинико-анамнестических данных. Как правило, выявляются случаи БТС у родственников, наличие периода нормального развития пациента, затем деградация – распад приобретенных навыков, сформировавшихся функций. Дифференциальная диагностика направлена на исключение дегенеративных заболеваний ЦНС, ювенильной идиотии, эпилепсии. Для подтверждения диагноза выполняются следующие процедуры:
Лечение болезни Тея-Сакса
В настоящее время эффективные способы терапии отсутствуют. Медицинская помощь больным нацелена на устранение симптомов и поддержание жизнедеятельности. Паллиативное лечение включает переход на зондовое питание, поскольку у пациентов утрачивается глотательный рефлекс, применение антибиотиков, противовирусных и иммуностимулирующих препаратов для борьбы с сопутствующими инфекционными заболеваниями. Терапия противоэпилептическими средствами не приносит положительного результата.
Поиск возможных способов лечения БТС продолжается. Исследования ведутся в трех направлениях: изучаются возможности ферментозаместительной, генной и субстратредуцирующей терапии. Замена отсутствующего фермента оказывается неэффективной из-за большого размера молекул гексозаминидазы, неспособных пройти сквозь гематоэнцефалический барьер и оболочку нейронов. Среди методов генотерапии опробовано введение нового генетического материала в клетки при помощи вирусного вектора и трансплантации стволовых клеток. Но положительных результатов не получено, исследования продолжаются. Наиболее перспективной считается субстратредуцирующая терапия с использованием фермента сиалидазы, который стимулирует катаболизм GM2 ганглиозидов. Ожидается разработка фармакологического препарата, повышающего экспрессию лизосомальных сиалидаз внутри нейронов.
Прогноз и профилактика
2. Клинический случай болезни Тея–Сакса с поздним началом/ Семенова О.В. и соавт.// Нервные болезни. - 2016 - №3.
Этиология и встречаемость болезни Тея-Сакса. Болезнь Тея-Сакса (MIM №272800), раннедетский ганглиозидоз GM2, — панэтническое аутосомно-рецессивное заболевание распада ганглиозидов, вызванное недостаточностью гексозаминидазы А (см. главу 12). Кроме раннедетской тяжелой формы, недостаточность гексозаминидазы А вызывает легкую форму болезни с началом в юношеском или взрослом возрасте.
Встречаемость недостаточности гексозаминидазы А широко варьирует в различных популяциях; встречаемость болезни Тея-Сакса в Северной Америке колеблется от 1 на 3600 новорожденных у евреев ашкенази до 1 на 360 000 среди не ашкенази евреев. Сравнимую с евреями-ашкенази встречаемость болезни Тея-Сакса имеют французские канадцы, каджуны в Луизиане и амиши в Пенсильвании. Повышенная частота носительства в этих четырех популяциях — следствие генетического дрейфа, хотя не исключено преимущество гетерозигот.
Патогенез болезни Тея-Сакса
Ганглиозиды — церамидовые олигосахариды, присутствующие в поверхностных мембранах всех клеток, но больше всего их в клетках мозга. Ганглиозиды концентрируются в поверхностных мембранах нейронов, особенно в аксонах и дендритах. Они функционируют как рецепторы различных гликопротеиновых гормонов и бактериальных токсинов и задействованы в дифференцировке клеток и межклеточном взаимодействии.
Гексозаминидаза А — лизосомный фермент, состоящий из двух субъединиц. а-Субъединица кодируется геном НЕХА в хромосоме 15, а бета-субъединица — геном НЕХВ в хромосоме 5. В присутствии белка-активатора гексозаминидаза А удаляет концевой N-ацетилгалактозамин из ганглиозида GM2.
Мутации генов а-субъединицы или белка-активизатора вызывают накопление GM2 в лизосомах и, этим самым, раннедетский, позднедетский или взрослый тип болезни Тея-Сакса. [Мутация а-субъединицы вызывает болезнь Сандхоффа (MIM № 268800)].
Механизм того, как накопление ганглиозида GM2 вызывает смерть нейронов, полностью не определен, хотя, по аналогии с болезнью Гоше, нейропатологию могут вызывать токсичные побочные продукты ганглиозида GM2. Уровень остаточной активности гексозаминидазы А обратно пропорционален тяжести болезни.
Пациенты с раннедетской формой ганглиозидоза GM2 имеют два патологических аллеля, приводящих к полному отсутствию активности гексозаминидазы. Пациенты с формами ганглиозидоза GM2 с началом в юношеском или взрослом возрасте — обычно сложные гетерозиготы по аллелю с полным отсутствием функции и аллелю с небольшой остаточной активностью гексозаминидазы А.
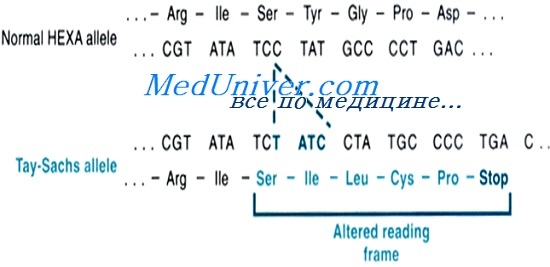
Фенотип и развитие болезни Тея-Сакса
Судороги обычно начинаются в конце первого года жизни и непрерывно становятся все тяжелее. Дальнейшее ухудшение на втором году жизни заканчивается децеребрационной ригидностью, затруднениями глотания, тяжелыми судорогами и, наконец, вегетативным состоянием.
Взрослый тип ганглиозидоза GM2 имеет выраженную клиническую изменчивость (прогрессирующая дистония, спиноцеребеллярная дегенерация, патология моторных нейронов или психиатрические нарушения). До 40% больных имеют прогрессирующие психиатрические проявления без психоза. Зрение затрагивается редко, и данные офтальмологического обследования обычно в норме.
Лечение болезни Тея-Сакса
Диагноз ганглиозидоза GM2 ставят на основании выявления как отсутствующей или почти отсутствующей активности гексозаминидазы А в сыворотке крови или в лейкоцитах, так и нормальной или повышенной активности гексозаминидазы В. Для диагностики также можно использовать анализ мутаций в гене НЕХА, но обычно его выполняют только для уточнения транспортного носительства и пренатальной диагностики.
Болезнь Тея-Сакса в настоящее время — инкурабельное заболевание; следовательно, лечение направлено на устранение симптомов и паллиативный уход. Почти все больные требуют фармакологического лечения судорог. Психиатрические проявления пациентов с взрослым типом ганглиозидоза GM2 обычно не поддаются стандартным антипсихотическим или противодепрессивным средствам; наиболее эффективны препараты лития и электросудорожная терапия.
Риски наследования болезни Тея-Сакса
Для потенциальных родителей без GM2-ганглиозидоза в семейном анамнезе эмпирический риск родить ребенка, больного СМ2-ганглиозидозом, зависит от частоты заболевания в их этнических группах. Для большинства жителей северной Америки эмпирический риск носительства составляет приблизительно 1 на 250-300, но для евреев-ашкенази эмпирический риск носительства — приблизительно 1 на 30. Для пары, в которой оба родителя носители, риск родить ребенка с ганглиозидозом GM2 равен 1/4.
Пренатальная диагностика основана на идентификации мутаций в гене НЕХА или на определении недостаточности гексозаминидазы А в тканях плода, например, ворсинах хориона или амниоцитах. Для эффективной идентификации пораженного плода с помощью анализа мутации в гене НЕХА обычно необходимо, чтобы мутации, вызывающие ганглиозидоз GM2 в семье, уже были известны.
Скрининг популяций высокого риска на носительство и последующие превентивные мероприятия уменьшили встречаемость болезни Тея-Сакса среди евреев-ашкенази почти на 90%. По традиции такой скрининг выполняют по определению активности гексозаминидазы А сыворотки крови с искусственным субстратом.
Этот чувствительный метод, тем не менее, не способен различить патологические мутации и псевдонедостаточность (снижение распада искусственного субстрата, но нормальный распад естественного субстрата); следовательно, носительство обычно подтверждают молекулярным анализом НЕХА. В гене НЕХА обнаружено два аллеля псевдонедостаточности и более 70 патологических мутаций.
Среди евреев-ашкенази, положительных по результатам ферментного скрининга, 2% — гетерозиготны по аллелю псевдонедостаточности, и 95-98% гетерозиготны по одной из трех патологических мутаций, две вызывают раннедетскую форму, одна — взрослую форму GM2-ганглиозидоза. В отличие от этого, среди остальных североамериканцев, положительных по результатам ферментативного скрининга, 35% — гетерозиготны по аллелям псевдонедостаточности.
Пример болезни Тея-Сакса. Семейная пара, оба евреи ашекенази, направлена в клинику генетики для оценки риска родить ребенка с болезнью Тея-Сакса. Сестра жены умерла от болезни Тея-Сакса в детстве. Дядя мужа по отцу находится в психиатрической лечебнице, но его диагноз неизвестен. Как муж, так и жена отказались от скрининга на носительство болезни Тея-Сакса в подростковом возрасте.
Анализ фермента показал, что как муж, так и жена имеют чрезвычайно низкую активность гексозаминидазы А. Последующий молекулярный анализ мутаций, преобладающих у евреев ашкенази, подтвердил, что у жены имеется патогенная мутация, тогда как у мужа только аллель псевдонедостаточности.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Мутация гена HEXA, ответственного за синтез фермента гексозоаминидазы A. Болезнь Тея-Сакса как редкое наследственное заболевание, которое передается ребенку от родителей. Формы болезни и характерный признак заболевания. Наследственное нарушение у детей.
| Рубрика | Медицина |
| Вид | реферат |
| Язык | русский |
| Дата добавления | 24.12.2013 |
| Размер файла | 12,3 K |
Студенты, аспиранты, молодые ученые, использующие базу знаний в своей учебе и работе, будут вам очень благодарны.
Введение
Болезнь Тея-Сакса - это тяжелое и редкое наследственное заболевание, которое передается ребенку от родителей и чаще всего имеет неутешительные прогнозы.
Болезнь впервые была описана в конце XIX века. Она получила свое название в честь британского офтальмолога Уоррена Тея и американского невролога Бернарда Сакса, которые внесли значительный вклад в изучение болезни. Выяснилось, что болезнь характерна для определенных этнических групп: чаще всего ею страдают евреи ашкеназы.
Причины заболевания
Болезнь вызывается мутацией гена HEXA, ответственного за синтез фермента гексозоаминидазы A - фермента, принимающего участие в метаболизме ганглиозидов. В результате, ганглиозиды накапливаются в нервных клетках, нарушая их работу.
Болезнью Тея-Сакса заболевает только ребенок, который унаследовал сразу два дефектных гена: от отца и матери. Если поврежденный ген есть только у одного из родителей, то ребенок, скорее всего, не заболеет, а окажется носителем, что рискованно для его потомства. Вероятность заболевания составляет 25%. На сегодняшний день известно более 100 различных мутаций в гене HEXA.
Формы болезни
Ранняя детская форма (инфантильный тип) начинается приблизительно в 6-месячном возрасте и характеризуется быстрым прогрессирующим течением: смерть наступает, как правило до 3-летнего возраста. Характерные признаки болезни- слепота с патогномоничной картиной глазного дна (вишнево-красное пятно в макулярной области, окруженное участком с белесоватым помутнением, и атрофия дисков зрительных нервов), прогрессирующее слабоумие и двигательные нарушения, в отдельных случаях - гиперкинезы, нистагм, снижение слуха, эпилептические припадки, эндокринные и трофические нарушения. В мозговой ткани значительно увеличено содержание ганглиозида типа Gm2, наиболее выраженное в белом веществе мозга. Накопление этого ганглиозида отмечено также в эритроцитах, селезенке и клетках печени. Основное значение в патогенезе инфантильного типа болезни Тея - Сакса придается дефициту фермента гексозаминидазы А, уровень которого снижен в мозге, внутренних органах и в крови. Исследование содержания гексозаминидазы А в крови используется для выявления гетерозиготных носителей патологического гена. Особенностью гемограммы больных является наличие вакуолизированных лимфоцитов.
Амавротическаяидиотия Тея - Сакса встречается чаще у детей еврейской национальности. Тип наследования аутосомно-рецессивный.
Поздняя детская форма начинается в возрасте около 3 лет и длится в среднем 6 лет. Характерные изменения на глазном дне отмечаются не у всех больных. В клинической картине наряду с прогрессирующим слабоумием и амаврозом на первый план выступают мозжечковые нарушения. Тип наследования аутосомно-рецессивный.
Ювенильная форма характеризуется началом в возрасте 5-8 лет, длительность болезни 10-15 лет. Отличается от предыдущих форм медленным прогрессированием и большим разнообразием клинической картины. Изменения психики не всегда достигают степени идиотии, прогрессирующее падение зрения также не всегда приводит к полной слепоте. Часто отмечаются эпилептические припадки, глухота, иногда глухонемота, ожирение. Из неврологических нарушений на первый план выступают экстрапирамидные, мозжечковые и бульбарные симптомы. Изменения на глазном дне характеризуются атрофией дисков зрительных нервов и признаками пигментного ретинита. В крови постоянно присутствуют вакуолизированные лимфоциты, количество которых в среднем составляет 21% от общего числа лимфоцитов. У гетерозиготов - 1% вакуолизированных лимфоцитов в крови. Тип наследования аутосомно-рецессивный.
Поздняя форма по срокам возникновения неоднородна. В большинстве семей заболевание начинается в юношеском возрасте или несколько позднее, характеризуется еще более медленным течением: больные доживают до 3 - 4-го десятилетия. Снижения интеллекта и зрения может быть выражено незначительно и даже отсутствовать. На глазном дне - картина пигментного ретинита. Часто отмечаются глухота, экстрапирамидные и мозжечковые расстройства, эпилептические припадки. Характерные патоморфологические изменения выражены лишь в отдельных участках и в значительно меньшей степени, чем при других формах амавротическойидиотии. В некоторых случаях отмечено экстранейрональное отложение жиросодержащего пигмента, отличающегося от пигмента, найденного при болезни Геллервордена - Шпатца.
Характерный признак заболевания - диффузная дегенерация ганглиозных клеток во всех отделах нервной системы. Процесс распада ганглиозных клеток и превращения многих из них в зернистую массу - шафферовская дегенерация - является патогномоничным признаком амавротическойидиотии. Отмечаются также распад миелиновых волокон, особенно в зрительных и пирамидных путях, дегенеративные изменения глии.
Заподозрить болезнь с самого начала у ребенка сложно. Обычно симптомы начинают проявляться с 4-6 месяцев:
ребенок слабо реагирует на свет и плохо фокусируется на предмете;
болезненно реагирует на шум, его пугает даже обычный голос человека;
при обследовании выявляются изменения на сетчатке глаза.
Второй момент проявления симптомов болезни - снижение двигательной активности ребенка с 6-месячного возраста. Он не может сидеть, плохо переворачивается, с ходьбой тоже возникают проблемы. В итоге развивается атрофия мышц или паралич, которые приводят к тому, что ребенок уже не может самостоятельно глотать и даже дышать.
Все это вместе с плохим зрением, слухом и их потерей впоследствии приводит к инвалидности. Голова становится несоразмерно большой. Между первым и вторым годом жизни часто наблюдаются судороги.
Если болезнь проявляется в раннем возрасте, ребенок обычно умирает до достижения 4-5 лет.
В другом случае заболевание может прогрессировать в период с 14 до 30 лет. У взрослых симптомы протекают легче:
может нарушаться речь;
страдает походка, координация и мелкая моторика;
наблюдаются мышечные спазмы;
ухудшается зрение, слух и интеллект.
Диагностика
Предположительный диагноз ставится после осмотра окулиста. При проверке органов зрения специалист обычно может обнаружить на глазном дне вишнево-красное пятно, что характерно для данного заболевания.
Далее подтвердить предположения помогает анализ на определение количества фермента в жидкостях и тканях исследуемого. Необходимы анализ крови и биопсия кожи. Если анализ положительный, это подтверждает диагноз либо носительство.
Определить, есть ли болезнь, до рождения ребенка, позволяет амниоцентез - анализ амниотической жидкости, полученной при проколе плодного пузыря.
Болезнь Тея-Сакса не поддается лечению. Клиническая картина обычно нарастает постепенно и также постепенно ведет к смерти.
Прогноз заболевания неутешителен: сначала болезнь ведет к инвалидности, а впоследствии к смерти. Продолжительность жизни больного зависит в первую очередь от тяжести симптомов заболевания. Бывает, что такие пациенты могут прожить столько же, сколько и здоровые люди.
Список литературы
Асанов А. Ю. с соавт. Основы генетики и наследственные нарушения у детей. М., 2003.
болезнь тей сакс
Подобные документы
Накопление липидов в лизосомах. Мутация гена, контролирующего синтез фермента 7-d-глюкоцереброзидазы. Нарушение функции макрофагов. Основные типы болезни Гоше. Клиническая картина ненейронопатического типа и нейронопатической инфантильной формы.
презентация [3,8 M], добавлен 08.03.2016
Наследственные болезни, связанные с нарушением липидного обмена: болезнь Гоше, болезнь Тей-Сакса, болезнь Ниманна-Пика. Симптоматика, течение болезни, методы диагностики, лечения и профилактики. Генетические аспекты заболеваний. Прогнозы для болеющих.
реферат [16,9 K], добавлен 06.01.2015
Болезнь Гентингтона как наследственное дегенеративное заболевание ЦНС, проявляющееся прогрессирующей хореей. Популяционная частота и этиология болезни. Формы ГАМК-эргического торможения. Классификация хореи Гентингтона по клиническим проявлениям.
реферат [1,1 M], добавлен 20.06.2009
Заболевание нервной системы. Сочетание прогрессирующего хореического гиперкинеза и психических расстройств. Первое описание симптомов болезни Гентингтона. Генетическое картирование (определение местонахождения) гена, ответственного за хорею Гентингтона.
презентация [2,3 M], добавлен 15.02.2017
Болезнь Паркинсона (паркинсонизм) как хроническое нейродегенеративное заболевание, его формы и основные симптомы болезней. Этиология и распространенность данного заболевания, механизм развития. Генетические и биохимические аспекты болезни Паркинсона.
Читайте также: