
Болезнь шарко мари тута реферат
Обновлено: 05.07.2024
Наследственные моторные и сенсорные невропатии (НМСН) являются наиболее распространенными дегенеративными поражениями периферической нервной системы, составляя приблизительно 40% хронических невропатий детского возраста (Ouvrier, 1992). Дегенерация миелиновых оболочек и/или аксонов вызывает преимущественно дистальную паралитическую амиотрофию, в основном поражающую нижние конечности и сопровождающуюся арефлексией. Моторное поражение затеняет сенсорные нарушения.
В основе действующей классификации наследственных моторных и сенсорных невропатий (НМСН) сохранились патоморфологические элементы (аксональная или демиелинизирующая), клинико-электрофизиологических проявлений (особенно моторная и сенсорная скорость проведения) и тип наследования. Пporpecc в молекулярной генетике быстро меняет критерии классификации. Многие из НМСН заканчиваются клинической картиной, называемой болезнь Шарко-Мари-Тута (ШМТ). Не так давно неврологи чаще использовали термин НМСН, тогда как генетики предпочитают ШМТ. Тип 1 НМСН синонимичен ШМТ 1, но есть некоторые несоответствия в номенклатуре, особенно в отношении более редких форм.
Например, ШМТ4 классифицирована как демиелинизирующая форма аутосомно-рецессивной НСМН, тогда как НСМНIV типа исходно соотносится с болезнью Рефсума. В данной главе будет использоваться преимущественно номенклатура ШМТ. В таблице ниже представлена комбинированная классификация.
В некоторых случаях клинические и даже морфологические (биопсия нерва) данные иногда минимальны, особенно у маленьких детей. Тем не менее дети, пораженные ШМТ 1 А, почти всегда демонстрировали некоторые симптомы, достигая возраста трех лет. Встречаются умеренные и даже бессимптомные случаи (Thomas et al., 1997), и поэтому систематическое обследование родителей и родственников улучшает диагностические возможности при предварительном консультировании. Большая часть дупликаций de novo имеет отцовское происхождение (Palau et al., 1993; Harding, 1995).
Болезнь Шарко-Мари-Тута (ШМТ) 1 является генетически гетерогенным типом, но более 80% случаев в детстве связаны с хромосомой 17 (ШМТ1А). Обычно дефект в таких случаях — субмикроскопическая дупликация, длиной приблизительно 1,5 мегабаз, в пределах участка 17р11.2 (Raeymaekers et al., 1991). Дупликация включает ген, кодирующий белок периферического миелина 22 (peripheral myelin protein 22 — РМР22) (Patel et al., 1992) так, чтобы ген был представлен в трех копиях. В нескольких случаях без дупликации была найдена точечная мутация гена РМР22 (Roa et al, 1993), что указывает на то, что этот ген — ген болезни при ШМТ 1.
В отдельных семьях болезнь вызывается мутациями в гене MPZ (myelin Р0) (ШМТ 1В), который картирован на участке хромосомы 1q21-q23 (Warner et al., 1996). Мутации генов, как РМР22, так и MPZ, вызывают разнообразные клинические картины, варьирующие в зависимости от локализации и тяжести мутации от врожденной гипомиелинизации, через фенотип Дежерина-Сотта к фенотипу ШМТ 1 или даже, в случае MPZ, до аксонального дегенеративного синдрома ШМТ (ШМТ2) (Warner et al., 1996). По нашему опыту педиатрических наблюдений, клинические проявления случаев, вызванных точечными мутациями, тяжелее, чем при типичных случаях ШМТ 1 А, связанных с дупликацией.
Некоторые случаи не картированы ни в одном из этих локусов. В некоторых доминантно наследуемых случаях, соотносимых с болезнью Шарко-Мари-Тута (ШМТ) 1C, выявлялись мутации гена LITAF/SIMPLE, который может быть вовлечен в аномальную деградацию белка (Street et al., 2003).
Х-сцепленная болезни Шарко-Мари-Тута (ШМТ X). Приблизительно в 10% случаев заболевание сцеплено с Х-хромосомой, где было выявлено по меньшей мере три локуса (Ionasescu et al., 1991, 1992). Самая частая (Х-сцепленная доминантная) форма находится на длинном плече на Xq13.1. Известно, что многочисленные мутации в этом очаге вовлекают ген коннексина 32 (Bergoffen et al., 1993), связывающего мембраны в зоне щелевых контактов белка, примыкающего к перехватам Ранвье и насечки Шмидта-Лантермана. Изучение родословных показывает ожидаемое отсутствие передачи от мужчины к мужчине. Обычно выраженность поражения у мужчин значительно выше, чем у женщин, но в целом клинические поражения в первое десятилетие менее тяжелы, чем при ШМТ 1 в том же возрасте. С другой стороны, возможная степень инвалидизации у взрослых мужчин больше при ШМТ X, чем при ШМТ 1.
Редкая Х-связанная рецессивная аксональная форма картирована на Хр22.2, и другая аксональная форма, связанная с глухотой и задержкой интеллектуального развития, картирована на Xq24-q26 (Priest et al., 1995; Ouvrier et al., 2007).
Невральная амиотрофия Шарко-Мари-Тута — это прогрессирующее хроническое наследственное заболевание с поражением периферической нервной системы, приводящем к мышечным атрофиям дистальных отделов ног, а затем и рук. Наряду с атрофиями наблюдается гипестезия и угасание сухожильных рефлексов, фасцикулярные подергивания мышц. К диагностическим мероприятиям относятся электромиография, электронейрография, генетическое консультирование и ДНК-диагностика, биопсия нервов и мышц. Лечение симптоматическое — курсы витаминотерапии, антихолинэстеразной, метаболической, антиоксидантной и микроциркуляторной терапии, ЛФК, массажа, физиопроцедур и водолечение.
МКБ-10

Общие сведения
Невральная амиотрофия Шарко-Мари-Тута (ШМТ) относится к группе прогрессирующих хронических наследственных полиневропатий, в которую входят синдром Русси-Леви, гипертрофическая невропатия Дежерина-Сотта, болезнь Рефсума и другие более редкие заболевания.
По различным данным, невральная амиотрофия Шарко-Мари-Тута встречается с частотой от 2 до 36 случаев на 100 тыс. населения. Зачастую болезнь носит семейный характер, причем у членов одной семьи клинические проявления могут иметь различную выраженность. Наряду с этим наблюдаются и спорадические варианты ШМТ. Лица мужского пола болеют чаще, чем женщины.
Причины
На сегодняшний день практическая неврология как наука не располагает достоверными сведениями о этиологии и патогенезе невральной амиотрофии. Проведенные исследования показали, что у 70-80% пациентов с ШМТ, прошедших генетическое обследование, отмечалось дублирование определенного участка 17-й хромосомы. Определено, что невральная амиотрофия Шарко-Мари-Тута имеет несколько форм, вероятно обусловленных мутациями различных генов. Например, исследователи выяснили, что при форме ШМТ, вызванной мутацией кодирующего митохондриальный белок гена MFN2, происходит образование сгустка митохондрий, нарушающего их продвижение по аксону.
Болезнь Шарко-Мари-Тута характеризуется аутосомно-доминантным наследованием с пенетрантностью на уровне 83%. Встречаются также случаи аутосомно-рецессивного наследования.
Патогенез
Установлено, что большинство форм ШМТ связаны с поражением миелиновой оболочки волокон периферических нервов, реже встречаются формы с патологией аксонов — осевых цилиндров проходящих в центре нервного волокна. Дегенеративные изменения затрагивают также передние и задние корешки спинного мозга, нейроны передних рогов, пути Голля (спинномозговые проводящие пути глубокой чувствительности) и столбы Кларка, относящиеся к заднему спинномозжечковому пути.
Вторично, в результате нарушения функции периферических нервов, развиваются мышечные атрофии, затрагивающие отдельные группы миофибрилл. Дальнейшее прогрессирование болезни характеризуется смещением ядер сарколеммы, гиалинизацией пораженных миофибрилл и интерстициальным разрастанием соединительной ткани. В последующем нарастающая гиалиновая дегенерация миофибрилл приводит к их распаду.
Классификация
В современной неврологической практике невральная амиотрофия Шарко-Мари-Тута подразделяется на 2 типа. Клинически они являются практически однородными, однако имеют ряд особенностей, позволяющих провести такое разграничение.
- Невральная амиотрофия I типа характеризуется существенным снижением скорости проведения нервного импульса. Биопсия нерва обнаруживает сегментарную демиелинизацию нервных волокон, гипертрофический рост непораженных шванновских клеток;
- При амиотрофии ШМТ II типа скорость проведения страдает незначительно, анализ биоптата показывает дегенерацию аксонов.
Отмечена связь болезни Шарко-Мари-Тута и атаксии Фридрейха. В отдельных случаях у пациентов с ШМТ со временем отмечаются типичные признаки болезни Фридрейха и наоборот — иногда по прошествии многих лет клиника атаксии Фридрейха сменяется симптоматикой невральной амиотрофии. Некоторыми авторами даны описания промежуточных форм этих заболеваний. Наблюдались случаи, когда у одних членов семьи диагностировалась атаксия Фридрейха, а у других — амиотрофия ШМТ.
Симптомы
В отдельных случаях невральная амиотрофия манифестирует расстройствами чувствительности в стопах, наиболее часто — парестезиями в виде ползания мурашек. Типичным ранним признаком ШМТ является отсутствие ахилловых, а позже и коленных сухожильных рефлексов. Основной симптом, на который пациенты чаще всего сами обращают внимание – приступообразные болезненные сокращения в икроножных мышцах (крампи), усиливающиеся в ночное время или после длительной физической нагрузки.
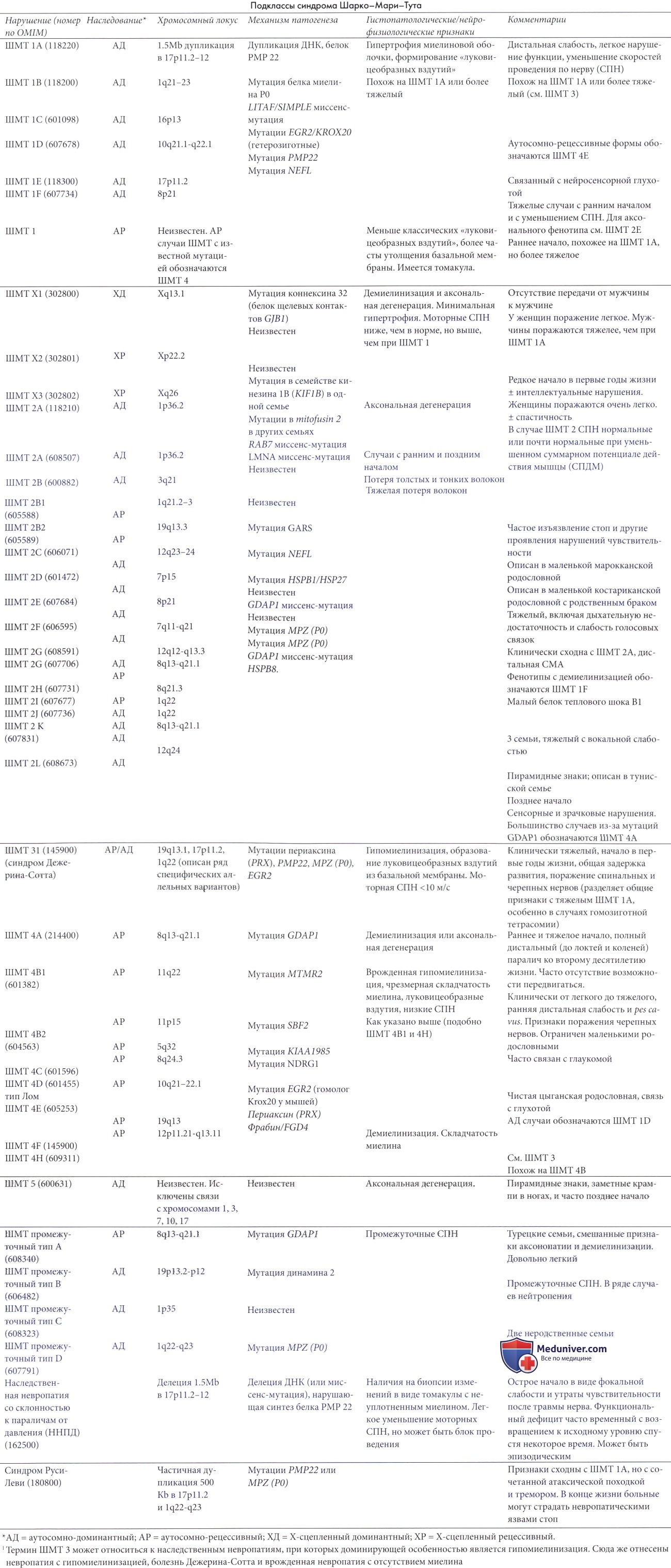
Развивающиеся первоначально атрофии затрагивают в первую очередь абдукторы и разгибатели стопы. Результатом является свисание стопы, невозможность ходьбы на пятках и своеобразная походка, напоминающая вышагивание лошади, — степпаж. Далее поражаются приводящие мышцы и сгибатели стопы. Тотальная атрофия мышц стопы приводит к ее деформации с высоким сводом, по типу стопы Фридрейха; формируются молоткообразные пальцы стопы. Постепенно атрофический процесс переходит на более проксимальные отделы ног — голени и нижние части бедер. В результате атрофии мышц голени возникает болтающаяся стопа. Из-за атрофии дистальных отделов ног при сохранности мышечной массы проксимальных отделов ноги приобретают форму перевернутых бутылок.
Зачастую при дальнейшем прогрессировании болезни Шарко-Мари-Тута атрофии появляются в мышцах дистальных отделов рук — вначале в кистях, а затем и в предплечьях. Из-за атрофии гипотенара и тенара кисть становиться похожей на обезьянью лапу. Атрофический процесс никогда не затрагивает мышцы шеи, туловища и плечевого пояса.
Часто невральная амиотрофия Шарко-Мари-Тута сопровождается легкими фасцикулярными подергиваниями мышц рук и ног. Возможна компенсаторная гипертрофия мышц проксимальных отделов конечностей. Сенсорные нарушения при невральной амиотрофии характеризуются тотальной гипестезией, однако поверхностная чувствительность (температурная и болевая) страдает значительно больше глубокой. В некоторых случаях наблюдается цианоз и отек кожи пораженных конечностей.
Для болезни Шарко-Мари-Тута типично медленное прогрессирование симптомов. Период между клинической манифестацией заболевания с поражения ног и до появления атрофий на руках может составлять до 10 лет. Несмотря на выраженные атрофии, пациенты длительное время сохраняют работоспособное состояние. Ускорить прогрессирование симптомов могут различные экзогенные факторы: перенесенная инфекция (корь, инфекционный мононуклеоз, краснуха, ангина, ОРВИ), переохлаждение, ЧМТ, позвоночно-спинномозговая травма, гиповитаминоз.
Осложнения
Невральная амиотрофия Шарко-Мари-Тута характеризуется ранней инвалидизацией. Вследствие прогрессирующей атрофии дистальных отделов конечностей и выраженных нарушений чувствительности больные постепенно теряют способность к самостоятельной ходьбе. Из-за грубых деформаций кистей рук пациенты не могут сами себя обслуживать. Контрактуры суставов нередко требуют хирургической коррекции.
На ранней стадии заболевания слабость в мышцах ног, гипестезия и гипорефлексия приводят к частым падениям, что повышает вероятность травм и переломов. Наиболее грозные неблагоприятные последствия происходят при сочетании болезни Шарко-Мари-Тута и атаксии Фридрейха. К ним можно отнести слепоту, кардиомиопатию, дыхательную недостаточность.
Диагностика
Курацией пациентов занимаются врачи-неврологи и ортопеды. При опросе больного уточняется возраст, в котором начали появляться симптомы (для болезни ШМТ типична манифестация в 15-25 лет). Важное значение имеет семейный анамнез (наличие близкого родственника с этой патологией). Во время общего осмотра обращается внимание на изменение походки, деформацию стоп и кистей.
При неврологическом осмотре отмечается уменьшение тонуса дистальных отделов верхних и нижних конечностей, ослабление или полное отсутствие сухожильных рефлексов (ахилловых, коленных), снижение кожной чувствительности. Для уточнения диагноза проводятся следующие методы исследования:
Дифференциальный диагноз невральной амиотрофии Шарко-Мари-Тута необходимо проводить с наследственными нейромышечными заболеваниями (спинальная мышечная атрофия Верднига-Гоффмана, адренолейкодистрофия, болезнь Пелицеуса-Мерцбахера) и приобретенными хроническими полинейропатиями (синдром Гийена-Барре).
Лечение невральной амиотрофии Шарко-Мари-Тута
Медикаментозная терапия
Для прохождения лечения все больные подлежат обязательной госпитализации в стационар. В настоящее время не существует специфической терапии, способной замедлить прогрессирование аксональной дегенерации и демиелинизации. Однако своевременно начатая грамотная и индивидуально подобранная терапия способна значительно улучшить качество жизни пациентов. Из лекарственных препаратов для симптоматического лечения невральной амиотрофии ШМТ применяются:
- Витамины. Для улучшения микроциркуляции и восстановления нервных волокон назначаются инъекции витаминов группы В (В1, В3, В12). К витамину В6 стоит относиться с осторожностью, так как превышение его дозы оказывает нейротоксический эффект. По данным некоторых исследователей, аскорбиновая кислота способна подавлять образование периферического белка миелина (PMP22).
- Миорелаксанты. С целью устранения болезненных мышечных сокращений пациентам рекомендуется прием медикаментов, расслабляющих скелетную мускулатуру – баклофен, толперизон.
- Кальций и витамин Д. Так как примерно 40% больных имеют остеопороз, для уменьшения риска переломов им показаны препараты кальция и витамина Д (холекальциферол).
- Антихолинэстеразные средства. При болезни ШМТ 2 типа для улучшения нервно-мышечной проводимости целесообразно назначение прозерина, галантамина.
Немедикаментозная терапия
Основное внимание уделяется немедикаментозному лечению невральной амиотрофии Шарко-Мари-Тута. Для достижения максимального терапевтического эффекта применяется комплекс следующих мероприятий:
- Электростимуляция. Для усиления нейротрофики, активации метаболизма в паретичных мышцах и проводимости периферических нервов используется направленная подача электрических импульсов.
- ЛФК. С целью повышения мышечного тонуса рекомендуются регулярные занятия лечебной физкультурой. Наиболее эффективно совмещение активных (выполняются самим пациентом) и пассивных (выполняются специалистом) упражнений.
- Массаж. Для улучшения кровообращения и лимфооттока в мышцах (в первую очередь нижних конечностей) выполняются различные виды массажа – ручной (стимулирующий, расслабляющий) и аппаратный (вибромассаж).
- Бальнеотерапия. Грязевые ванны и грязевые аппликации способствуют коррекции нарушений вегетативной нервной системы и замедлению формирования контрактур.
- Ортопедическое лечение. Чтобы предупредить развитие грубых деформаций больным назначается ношение ортопедической обуви. При нестабильности суставов из-за мышечной слабости, для фиксации стоп в заданном положении используются специальные приспособления (ортезы, подтяжки).
Хирургическое лечение
При выраженных атрофических явлениях и деформации стопы, значительно затрудняющих самостоятельную ходьбу, когда консервативные методы оказываются безуспешными, показаны ортопедические оперативные вмешательства – метатарзальная остеотомия, остеотомия пяточной кости. В некоторых случаях для восстановления опорной функции стопы может понадобиться проведение артродеза.
Экспериментальное лечение
Продолжаются поиски эффективного лекарства для борьбы с невральной амиотрофией Шарко-Мари-Тута. В клинических испытаниях, где пациенты принимали препарат PXT3003 (комбинация малых доз баклофена, налтрексона и сорбитола), были отмечены положительные результаты в виде увеличения мышечной силы, возобновления чувствительности и сухожильных рефлексов.
Рассматривается возможность использования в качестве лечения ингибиторов HDAC6 – ферментов, стимулирующих регенерацию белков цитоскелета нервных клеток. Эксперименты на лабораторных животных показали, что данные вещества способны значительно замедлить прогрессирование демиелинизации и аксональной дегенерации.
Прогноз и профилактика
Невральная амиотрофия Шарко-Мари-Тута – тяжелое инвалидизирующее заболевание. Большинство пациентов утрачивают способность ходить через 15-20 лет после начала появления симптомов. Однако в виду того, что преимущественно поражаются дистальные отделы конечностей, продолжительность жизни больных практически не отличается от таковой в общей популяции.
Летальные исходы в молодом и среднем возрасте наблюдаются при сочетании с атаксией Фридрейха, когда в патологический процесс вовлекается дыхательная мускулатура и миокард. Специфических методов первичной профилактики не существует. Предупредить развитие осложнений и максимально сохранить работоспособность позволяет своевременное начало комплексной терапии.
1. Оценка качества жизни больных с наследственной невропатией Шарко—Мари—Тута в Красноярском крае/ Шнайдер Н.А., Глущенко Е.В., Козулина Е.А.// Бюллетень Сибирской медицины. - 2011.
2. Клинико-генетическая характеристика наследственной невропатии Шарко—Мари—Тута (на примере Красноярского края): Автореферат диссертации/ Глущенко Е.В. - 2011.
3. Ведение и реабилитация пациентов с наследственной невропатией Шарко—Мари—Тута/ Шнайдер Н.А., Глущенко Е.В.// Комплексная реабилитация: наука и практика. - 2010.
4. Наследственная невропатия Шарко—Мари—Тута/ Шнайдер Н.А., Глущенко Е.В., Кантимирова Е.А. и др. - 2010.
Говоря о полой стопе нельзя пройти мимо основной наследственной причины этой патологии – болезни Шарко Мари Тута. Другое её название - наследственная моторная сенсорная нейропатия, различают две формы первая с превалированием мышечной слабости, вторая приводящая к нарушению чувствительности. Является наиболее частым наследственным неврологическим заболеванием (частота 1:2500). При этом заболевании нарушается иннервация таких периферических мышц как: короткая малоберцовая мышца, передняя большеберцовая мышца, собственные мышцы стопы и кисти.

Своим витиеватым названием наследственная моторная сенсорная нейропатия обязана трём врачам: Jean-Martin Charcot (1825–1893), Pierre Marie (1853–1940) Howard Henry Tooth (1856–1925). Их фамилии и легли в основу названия болезни Charcot – Marie – Tooth.
Болезнь Шарко Мари Тута чаще наследуется по аутосомно-доминантному принципу. Это значит, что 50 % детей наследуют заболевание от больного родителя. Также встречается аутосомно-рецессивный тип наследования и сцепленный с X-хромосомой.
Таким образом можно выделить две основные формы болезни, в первом случае происходит просто нарушение функции нервов, во втором их дегенерация.
Ортопедические проявления болезни Шарко Мари Тута – полая стопа, молоткообразные пальцы, дисплазия тазобедренных суставов, сколиоз.
Классификация болезни Шарко Мари Тута.
I А тип болезни ШМТ – демиелинизирующее заболевание, сопровождающееся значительным снижением скорости проведения нервных импульсов. Аутосомно-доминантное заболевание, проявления появляются в первой-второй декаде жизни, чаще всего приводит к формированию полой стопы.
II тип ШМТ – постепенное отмирание аксонов за счёт их дегенерации. Имеет более мягкое течение, начинается во второй-третей декаде жизни, чаще приводит к плоской стопе.
Симптомы болезни Шарко Мари Тута.
Снижение чувствительности к касанию в стопах, голеностопе и голени постепенно прогрессирует, сопровождается болезненными мышечными спазмами, интенсивность которых значительно варьирует в зависимости от активности процесса. Физическая нагрузка часто провоцирует онемение, спазмы и боль в стопах и кистях. В более тяжёлых случаях болезнь может затрагивать челюстные мышцы, голосовые связки, околопозвоночные мышцы, сопровождаясь нарушениями произношения и сколиозом. Стресс, беременность, длительная иммобилизация приводят к обострению заболевания.
Суммируя наиболее часто встречаемые симптомы: боль по наружному краю стопы, снижение тактильной чувствительности, хромота, частые травмы голеностопного сустава, проблемы с ходьбой по лестнице.
Диагностика болезни Шарко Мари Тута.
При физическом осмотре в первую очередь обращают внимание на деформацию стопы.
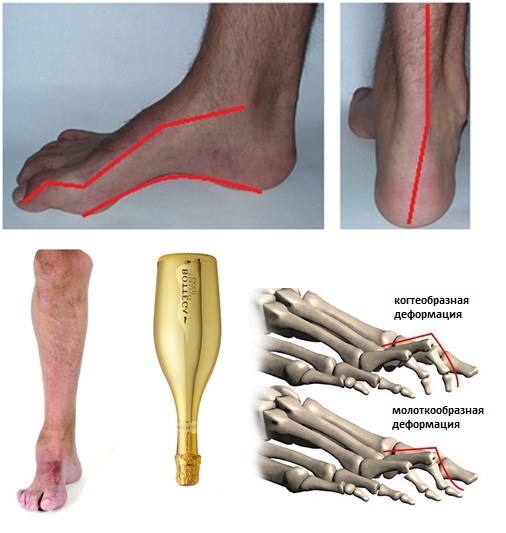
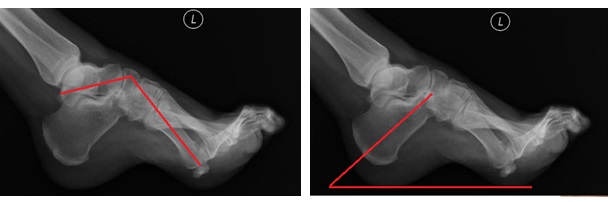
Первоначально определяется смещение первого луча в подошвенную сторону. Вторым элементом появляется кавусная деформация за счёт того, что длинная малоберцовая мышца (нормальная) оказывается сильнее поражённой передней большеберцовой мышцы. Третьим элементом деформации становится варусная деформация, которая появляется за счёт того, что задняя большеберцовая мышца оказывается сильнее поражённой короткой малоберцовой мышцы.
Вторым важным моментом является определение атрофии короткого разгибателя пальцев и короткого разгибателя большого пальца стопы, атрофии голени, особенно в нижнем её отделе, слабость тыльной флексии и эверсии стопы, снижение рефлексов нижней конечности.
Также выполняется тест Колмана (Coleman block test) для определения эластичности заднего отдела стопы.
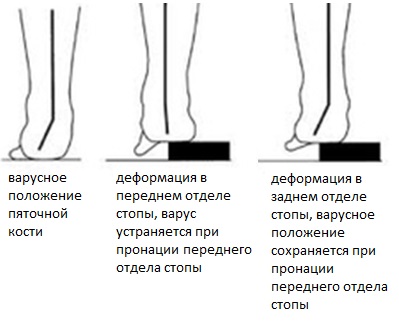
При подкладывании дощечки под наружный край стопы при элдастичной деформации заднего отдела происходит её коррекция до нейтрального положения, в случае ригидной деформации положение пяточной кости остаётся неизменным.
При осмотре верхних конечностей отмечается атрофия собственных мышц кисти.
ЭМГ – снижение скорости проведения нервного возбуждения в малоберцовом, локтевом, срединном нервах.
Генетическое обследование – ПЦР для определения мутации в гене PMP 22, хромосомный анализ для выявлении дупликации плеча 17 хромосомы при самой распространённой аутосомно-доминантной форме заболевания.
С принципами консервативного и хирургического лечения деформации стопы при болезни Шарко Мари Тута вы можете ознакомится в статье посвящённой полой стопе.

Никифоров Дмитрий Александрович
Хирургия стопы и голеностопного сустава, коррекция деформаций конечностей, эндопротезирование суставов, артроскопическая хирургия, спортивная травма.
Частота диагностирования болезни Шарко-Мари-Тута согласно статистическим данным где-то один случай на две с половиною тысячи пациентов. Первые симптомы появляются в молодом возрасте. Выраженность симптомов и скорость прогрессирования болезни Шарко-Мари разная у каждого пациента. Процент инвалидизации при заболевании очень высок.

Причины заболевания болезнью Шарко:
- Мутация гена PMP22;
- Мутация MPZ;
- Мутация GJB1;
- Мутация MFN и др.
- аутосомно-доминантный (чаще всего);
- аутосомно-рецессивный;
- Х-сцепленный.
Заболевание имеет много форм, вызванные разным видом мутаций. Качество жизни и возможности работоспособности при болезни Шарко-Мари-Тута значительно ухудшаются, но на продолжительности жизни обычно это не сказывается.
Симптомы болезни Шарко связанные с поражением моторных и сенсорных нервных волокон. Диагностика болезни Шарко-Мари заключается в исключении диагнозов, которые могут давать подобную клиническую картину, и в проведении ДНК-диагностики, но учитывая, что не все виды мутаций известны, она не всегда информативна.
Лечение болезни Шарко-Мари-Тута заключается в симптоматической терапии. Специфическое лечение на данный момент все еще находится в стадии разработки.
Юсуповская больница – одно из лучших медицинских учреждений, где лечатся пациенты с болезнью Шарко-Мари-Тута. Несмотря на то, что терапия направлена только на купирование симптомов, неврология стремительно развивается и питается найти способы лечения многих заболеваний, в том числе и болезни Шарко. Ведение пациентов изданной патологией достаточно сложное, ведь клиническая картина разнообразна, симптомы выражены в неодинаковой степени и п.т. Опыт работы специалистов Юсуповской больницы позволяет оказывать качественную и эффективную медицинскую помощь. Доктора следят за клиническими исследованиями, новыми разработками, препаратами, изучают их эффективность. В случае необходимости, диагностика проводится быстро и с использованием нового оборудования. Персонал работает на благо пациента.
Симптомы болезни Шарко
Симптомы болезни Шарка-Мари появляются в молодом возрасте, чаще всего до двадцати лет. Прогрессирует заболевание постепенно, пациенты долгое время сохраняют работоспособность и возможность самообслуживания. Причинами, которые ускоряют развитие заболевания, могут стать вирусные и бактериальные инфекции, воздействия неблагоприятных факторов среды, травматизм, недостаток витаминов и т.п.
Дальнейшее развитие заболевания болезни Шарко-Мари-Тута приводит к втягиванию в процесс и кистей, далее - предплечий. Мышцы шеи, туловища и плечевого пояса не атрофируются. Мышцы проксимальных отделов компенсаторно увеличиваются.
Все виды чувствительности нарушаются, но больше всего страдает поверхностная, особенно температурная.
Диагностика болезни Шарко
- Осмотр неврологом;
- Лабораторные исследования;
- Инструментальные исследования;
- ДНК-исследования.
Полное обследование необходимо для исключения заболеваний, в которых клиническая картина сходна. К ним относят: боковой амиосклероз, миотония, метаболическая невропатия. Для исключения хронических полинейропатий проводят биопсию мышц.
Лечение болезни Шарко
Все способы лечения болезни Шарко-Мари-Тута не радикальны. Симптоматическое лечение включает медикаментозную терапию, физиотерапию, лечение у ортопеда и т.п.
Физиотерапевтическое лечение болезни Шарко-Мари-Тута включает ЛФК, массаж, электрофорез, диадинамотерапию, терапию лечебными грязями, разные виды ванн и др.
Медикаментозная терапия направлена на улучшение питания мышечных волокон. С этой целью назначают кокарбоксилазу, глюкозу, аденозинтрифосфат и др. Так же широко применяют антиоксидантные средства, препараты, улучшающие микроциркуляцию, и витамины. Хорошо зарекомендовали себя препараты, которые тормозят активность ацетилхолинэстеразы и повышают уровень ацетилхолина, например, прозерин, галантамин.
Дальнейшие разработки новых препаратов, направленные на радикальные меры – это мир без болезни Шарко-Мари-Тута. Прогрессирование болезни Шарко-Мари-Тута не отражается на том, сколько живут пациенты.
В Юсуповской больнице специалисты долгие годы помогают пациентам держать болезнь под контролем. Минимальная выраженность симптомов и медленное прогрессирование – результат работы врачей. В комфортных палатах, на новых тренажерах, в хорошо оснащенных кабинетах – вот где проходит лечение болезни Шарко-Мари-Тута. Не затягивайте с лечением, запишитесь на консультацию.
Читайте также: