
Болезнь минковского шоффара реферат
Обновлено: 02.07.2024
Что такое Наследственный сфероцитоз (болезнь Минковского-Шоффара) -
Наследственный сфероцитоз (болезнь Минковского-Шоффара) - гемолитическая анемия вследствие дефекта клеточной мембраны эритроцитов, проницаемость мембраны для ионов натрия становится избыточной, в связи с чем эритроциты приобретают шарообразную форму, становятся ломкими и легко подвергаются спонтанному гемолизу.
Наследственный сфероцитоз является широко распространенным заболеванием (2-3 случая на 10000 населения) и встречается у лиц большинства этнических групп, однако чаще болеют жители северной части Европы.
Что провоцирует / Причины Наследственного сфероцитоза (болезни Минковского-Шоффара):
Наследственный сфероцитоз передается по аутосомно-доминантному типу. Как правило, у одного из родителей выявляют признаки гемолитической анемии. Возможны спорадические случаи заболевания (в 25%), представляющие собой новые мутации.
Патогенез (что происходит?) во время Наследственного сфероцитоза (болезни Минковского-Шоффара):
В патогенезе наследственного сфероцитоза бесспорны 2 положения: наличие генетически детерминированной аномалии белков, или спектринов, мембраны эритроцитов и элиминирующая роль селезенки в отношении сфероидальноизмененных клеток. У всех больных с наследственным сфероцитозом отмечен дефицит спектринов в эритроцитарной мембране (до 1/3 нормы), а у некоторых - нарушение их функциональных свойств, причем установлено, что степень дефицита спектрина может коррелировать с тяжестью заболевания.
Наследственный дефект структуры мембраны эритроцитов приводит к повышенной проницаемости ее для ионов натрия и накоплению воды, что в свою очередь ведет к чрезмерной метаболической нагрузке на клетку, потере поверхностных субстанций и формированию сфероцита. Формирующиеся сфероциты при движении через селезенку начинают испытывать механическое затруднение, задерживаясь в красной пульпе и подвергаясь всем видам неблагоприятных воздействий (гемоконцентрация, изменение рН, активная фагоцитарная система), т.е. селезенка активно наносит сфероцитам повреждения, вызывая еще большую фрагментацию мембраны и сферуляцию. Это подтверждается при электронно-микроскопических исследованиях, позволивших обнаружить ультраструктурные изменения в эритроцитах (утолщение клеточной мембраны с ее разрывами и образованием вакуолей). Через 2-3 пассажа через селезенку сфероцит подвергается лизису и фагоцитозу. Селезенка является местом гибели эритроцитов; продолжительность жизни которых сокращается до 2 недель.
Хотя дефекты эритроцитов при наследственном сфероцитозе обусловлены генетически, в организме возникают условия при которых углубляются эти дефекты и реализуется гемолитический криз. Кризы могут провоцироваться инфекциями, некоторыми химическими веществами, психическими травмами.
Симптомы Наследственного сфероцитоза (болезни Минковского-Шоффара):
Наследственный сфероцитоз может проявляться с неонатального периода, однако более выраженные симптомы обнаруживают к концу дошкольного и в начале школьного возраста. Раннее проявление заболевания предопределяет более тяжелое течение. Чаще болеют мальчики.
Наследственный сфероцитоз - гемолитическая анемия с преимущественно внутриклеточным типом гемолиза, это обусловливает и клинические проявления болезни - желтуху, увеличение селезенки, большую или меньшую степень анемии, склонность к образованию камней в желчном пузыре.
Жалобы, клинико-лабораторная симптоматика во многом определяются периодом заболевания. Вне гемолитического криза жалобы могут отсутствовать. При развитии гемолитического криза отмечаются жалобы на повышенную утомляемость, вялость, головную боль, головокружение, бледность, желтуху, снижение аппетита, боли в животе, возможны повышение температуры до высоких цифр, тошнота, рвота, учащение стула, грозный симптом - появление судорог.
Симптоматика криза во многом определяется анемией и зависит от степени гемолиза.
При объективном обследовании кожа и видимые слизистые бледные или лимонно-желтые. У детей с ранними проявлениями наследственного сфероцитоза возможны деформации скелета, особенно черепа (башенный, квадратный череп, изменяется расположение зубов и т.д.); нередки генетические стигмы. У больных обнаруживаются разной степени выраженности изменения со стороны сердечно-сосудистой системы, обусловленные анемией. Характерен гепатолиенальный синдром с преимущественным увеличением селезенки. Селезенка плотная, гладкая, нередко болезненная, что, по-видимому, объясняется напряжением капсулы вследствие кровенаполнения или периспленитом. Окраска экскрементов в момент криза интенсивная. Следует отметить возможные колебания в размерах селезенки: значительное увеличение при гемолитических кризах и уменьшение в период относительного благополучия.
В зависимости от тяжести наследственного сфероцитоза клинические симптомы могут быть выражены незначительно. Иногда желтуха может быть единственным симптомом, по поводу которой больной обращается к врачу. Именно к этим лицам относится известное выражение Шоффара: "Они более желтушны, чем больны." Наряду с типичными классическими признаками заболевания встречаются формы наследственного сфероцитоза, когда гемолитическая анемия может быть настолько хорошо компенсирована, что пациент узнает о заболевании лишь при проведении соответствующего обследования.
Наряду с наиболее типичными гемолитическими кризами при тяжелом наследственном сфероцитозе возможны арегенераторные кризы с симптомами гипоплазии преимущественно красного ростка костного мозга. Такие кризы могут развиваться остро с довольно яркими симптомами анемии-гипоксии и наблюдаются обычно у детей после 3 лет жизни. Арегенераторные кризы кратковременны (1-2 недели) и носят обратимый характер в отличие от истинной аплазии.
Наследственный сфероцитоз осложняется образованием пигментных камней в желчном пузыре и желчных протоках, после 10 лет камни желчного пузыря встречаются у половины больных, не подвергнутых спленэктомии.
Диагностика Наследственного сфероцитоза (болезни Минковского-Шоффара):
Диагноз наследственный сфероцитоз ставится на основании генеалогического анамнеза, клинических данных, описанных выше и лабораторных исследований. Гемолитическую природу анемии подтверждают нормохромная нормоцитарная анемия с ретикулоцитозом, непрямая гипербилирубиннемия, степень выраженности которых зависит от тяжести гемолиза. Окончательный диагноз основывается на морфологических особенностях эритроцитов и характерном признаке наследственного сфероцитоза - изменении осмотической резистентности эритроцитов.
Течение наследственного сфероцитоза волнообразное. Вслед за развитием криза улучшаются клинико - лабораторные показатели и наступает ремиссия, которая может длиться от нескольких недель до нескольких лет.
Дифференциальный диагноз. Наследственный сфероцитоз следует дифференцировать от других врожденных гемолитических анемий. Данные семейного анамнеза, исследование мазков крови и осмотической резистентности эритроцитов отличаются наибольшей диагностической ценностью.
Из других заболеваний наследственный сфероцитоз прежде всего дифференцируют с гемолитической болезнью новорожденных, в более старшем возрасте - с вирусным гепатитом, аутоиммунной гемолитической анемией.
Лечение Наследственного сфероцитоза (болезни Минковского-Шоффара):
Радикальным методом лечения наследственного сфероцитоза является спленэктомия, которая обеспечивает практическое выздоровление, несмотря на сохранность сфероцитов и снижение осмотической резистентности (степень их выраженности уменьшается). Оптимальный возраст для проведения операции 5-6 лет. Однако возраст не может рассматриваться как противопоказание к хирургическому лечению. Тяжелые гемолитические кризы, их непрерывное течение, арегенераторные кризы - показания для проведения спленэктомии даже у детей раннего возраста. Существует повышенная склонность к инфекционным заболеваниям в течение 1 года после операции. В связи с этим в ряде стран принято ежемесячное введение в течение одного года после спленэктомии бициллина - 5 или перед плановой спленэктомией проводят иммунизацию пневмококковой поливакциной.
Прогноз при наследственном сфероцитозе благоприятный. Однако, в тяжелых случаях гемолитического криза при несвоевременном лечении является серьезным (возможен летальный исход).
Так как наследственный сфероцитоз наследуется по аутосомно-доминантному типу с довольно высокой пенетрантностью гена, необходимо учитывать, что степень риска рождения больного ребенка (любого пола) при наличии наследственного сфероцитоза у одного из родителей составляет 50%. Дети, больные наследственным сфероцитозом, находятся на постоянном диспансерном учете.
Диета. Введение в рацион повышенного количества фолиевой кислоты (более 200 мкг/сут). Рекомендуемые продукты: хлебобулочные изделия из муки грубого помола, гречневая и овсяная крупы, пшено, соя, фасоль, измельченные сырые овощи (цветная капуста, зелёный лук, морковь), грибы, говяжья печень, творог, сыр.
Профилактика Наследственного сфероцитоза (болезни Минковского-Шоффара):
Наследственный сфероцитоз предупредить невозможно. Однако люди, больные наследственным сфероцитозом, могут обратиться к консультанту-генетику с тем, чтобы обсудить возможности определения дефектного гена, вызываю¬щего заболевание их детей.
Профилактика наследственного сфероцитоза сводится к лечебным мероприятиям во время кризов.
К каким докторам следует обращаться если у Вас Наследственный сфероцитоз (болезнь Минковского-Шоффара):
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Наследственного сфероцитоза (болезни Минковского-Шоффара), ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору .
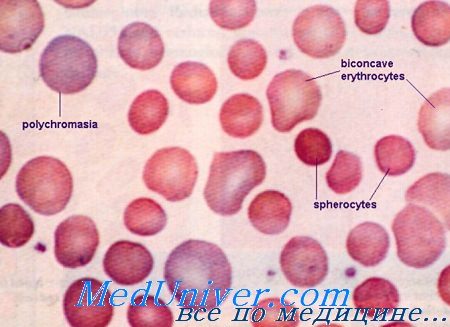
Наследственный микросфероцитоз – это гемолитическая анемия, обусловленная генетическим дефектом мембран эритроцитов и характеризующаяся постоянным гемолизом. Клинические признаки включают бледность, желтушность кожи, слизистых, боли в левой части живота за счет увеличения селезенки, а также деформацию скелета. В раннем возрасте развивается желчнокаменная болезнь. Диагностика осуществляется с помощью общего анализа крови, определения осмотической резистентности эритроцитов. Иногда требуется проведение электрофореза мембранных белков. Основным лечением является удаление селезенки (спленэктомия).
МКБ-10


Общие сведения
Наследственный микросфероцитоз (НМС, болезнь Минковского-Шоффара) – врожденное гематологическое заболевание из группы мембранопатий. Впервые болезнь подробно была описана немецким терапевтом Оскаром Минковским в 1900 г., спустя 7 лет французский терапевт Анатоль Шоффар установил снижение осмотической резистентности красных кровяных телец при НМС. Распространенность данной патологии в среднем составляет 1: 2500 человек, несколько чаще она встречается в Японии, странах Африки, Северной Европы. Клиническая манифестация может произойти в любом возрасте, но чаще наступает в юношеском или зрелом возрасте. Больше страдают лица мужского пола.
Причины
В основе наследственного микросфероцитоза лежит мутация гена, кодирующего синтез одного из белков цитоскелета мембраны эритроцитов. В разных семьях обнаруживаются мутации различных генов. Ген альфа-цепи спектрина расположен на 1 хромосоме (локус Iq21), ген бета-цепи – на 14 хромосоме (локус q22-q23), а ген анкирина – на 8 хромосоме (локус 8p 11.2). Болезнь характеризуется аутосомно-доминантным типом наследования.
К предрасполагающим факторам можно отнести наличие среди близких родственников больного наследственным микросфероцитозом или бессимптомного носителя мутантных генов. Спровоцировать резкое обострение (гемолитический криз) или первое проявление НМС у лиц с легкой формой могут инфекционные патологии, вакцинация, сильный стресс. У женщин обострения нередко возникают при наступлении беременности.
Патогенез
Вследствие постоянной деструкции красных клеток компенсаторно усиливаются процессы костномозгового кроветворения. Из-за хронического гемолиза высвобождается большое количество неконъюгированного билирубина, который направляется в печень для секреции в желчь. Поэтому желчный пузырь начинает заполняться пигментными камнями. При патологоанатомическом исследовании обнаруживают гиперплазию эритроидного ростка костного мозга трубчатых, плоских костей. Кровенаполнение пульпы резко выражено. Также нередко отмечается гемосидероз внутренних органов.
Классификация
Яркость клинической картины зависит от того, дефицит какого белка наблюдается у пациента, и является он гетерозиготным или гомозиготным носителем мутантных генов. По этим критериям различают следующие степени тяжести наследственного микросфероцитоза:
- Легкая. Небольшой гемолиз, развивающийся у взрослых людей под влиянием провоцирующих факторов. Селезенка увеличена незначительно. Уровень гемоглобина 100-120 г/л.
- Средняя. Умеренный гемолиз и спленомегалия. Кожа желтушной окраски. Уровень гемоглобина 80-100 г/л.
- Тяжелая. Редкая форма. Выраженный гемолиз, большие размеры селезенки, скелет деформирован. Характерно кризовое течение с большим количеством осложнений и вероятностью летального исхода. Уровень гемоглобина 60-80 г/л. Имеется потребность в постоянных гемотрансфузиях.
- Бессимптомная (латентная). При этой разновидности человек даже не подозревает, что болен. Данная форма характерна для гетерозиготных лиц. Единственным признаком может быть наличие небольшого количества микросфероцитов, высокий ретикулоцитоз. Истинная частота распространенности неизвестна.
Симптомы наследственного микросфероцитоза
Начало заболевания обычно постепенное. При латентной и легкой форме усиленное костномозговое кроветворение компенсирует постоянное разрушение эритроцитов, что позволяет поддерживать уровень гемоглобина на должном уровне. Тяжесть клинической картины определяется степенью гемолиза. На первый план обычно выступает желтушность кожных покровов, склер, слизистой оболочки рта с лимонно-шафрановым оттенком. Долгое время желтуха может быть единственным признаком наследственного микросфероцитоза.
Анемичный синдром проявляется бледностью кожи, слизистых, симптомами пониженного артериального давления (слабостью, головокружением, тахикардией). Практически всегда увеличена селезенка, из-за чего больной испытывает тянущую или ноющую боль в левом подреберье. Желтуха, спленомегалия, анемия составляют гемолитическую триаду. Нередко увеличена печень, но не так сильно, как селезенка, поэтому тяжесть и боль в правом подреберье незначительны.
Если заболевание манифестирует с раннего детского возраста, то развивается деформация костного скелета (стигмы дизэмбриогенеза) – башенный череп, укорочение мизинцев, широкая переносица и т. д. У взрослых больных с тяжелой формой НМС, которым не была проведена спленэктомия, иногда наблюдаются трофические язвы нижних конечностей (область голени, лодыжек), что обусловлено ухудшением микроциркуляции.
Особо яркую клинику имеет гемолитический криз, возникающий под влиянием различных провоцирующих факторов. Вследствие массивного гемолиза у больного повышается температура тела, нарастает интенсивность желтухи. Присоединяются симптомы билирубиновой интоксикации (потеря аппетита, рвота, боли в мышцах, суставах). Из-за резкого увеличения селезенки боли усиливаются, приобретают распирающий характер. Уровень гемоглобина падает до критических значений, пациент может потерять сознание.
Осложнения
Наиболее частыми осложнениями (50%) считаются желчнокаменная болезнь и калькулезный холецистит, возникающие по причине высвобождения из разрушенных эритроцитов большого количества билирубина, секретирующегося в желчь. Длительные переливания крови могут привести к перегрузке железом, вторичному гемохроматозу (цирроз печени, кардиомиопатия, сахарный диабет 2 типа). Трофические язвы ног в редких случаях способствуют развитию бактериальных воспалительных процессов в подкожной, межмышечной клетчатке (флегмона, некротизирующий фасциит).
Самое опасное состояние, которое возникает при наследственном микросфероцитозе, – апластический криз, вызванный инфицированием парвовирусом В19. В костном мозге прекращаются процессы кроветворения, резко снижается содержание в крови всех форменных элементов (эритроцитов, лейкоцитов, тромбоцитов). Появляются кровотечения, глубокая анемия, высокая чувствительность к инфекционным агентам.
Диагностика
Пациенты с болезнью Минковского-Шоффара подлежат обследованию у врача-гематолога. При осмотре пациента учитывается степень выраженности гемолитической триады, наличие деформации лицевого скелета. Уточняется, есть ли данное заболевание у кого-либо из близких родственников. С целью подтверждения диагноза назначается дополнительное обследование, которое включает:
- Анализы крови. В общем анализе крови обнаруживаются снижение уровня эритроцитов, гемоглобина, увеличение ретикулоцитов (до 20%). Размер эритроцитов уменьшен (меньше 7 мкм). При морфологической оценке мазка крови выявляются микросфероциты. Биохимический анализ крови показывает признаки гемолиза – высокую концентрацию непрямого билирубина, лактатдегидрогеназы.
- Верифицирующие тесты. Выявляется уменьшение осмотической устойчивости эритроцитов в виде повышенной чувствительности к лизису в гипотонических растворах NaCl (0,4-0,6%). Также характерна слабая способность красных клеток крови к фиксации флуоресцентного красителя эозин-5-малеимида при ЭМА-тесте. Электрофорез мембранных белков эритроцитов позволяет точно определить дефицит конкретного протеина (спектрин, анкирин).
- Инструментальные исследования. При УЗИ органов брюшной полости у пациента с любой формой наследственного микросфероцитоза обнаруживается увеличение селезенки, а иногда и печени. Часто находят камни в желчном пузыре. На рентгенографии костей черепа, трубчатых костей видны признаки разрастания костного мозга – расширение костномозгового канала, участки остеопороза, истончение кортикального слоя.
Спектр исключаемых патологий при болезни Минковского-Шоффара довольно широк. Наиболее часто приходится дифференцировать НМС от аутоиммунных гемолитических анемий. С этой целью для исключения иммунной природы гемолиза проводят антиглобулиновый тест (реакцию Кумбса). При НМС результат отрицательный. Легкие формы, сопровождающиеся лишь желтухой и небольшой спленомегалией, нужно отличать от доброкачественных гипербилирубинемий (синдрома Жильбера). Перегрузку железом дифференцируют с первичным (наследственным) гемохроматозом.
Лечение наследственного микросфероцитоза
Пациенты с легкой и бессимптомной формой не нуждаются в лечении. Им необходимо лишь регулярно посещать врача, сдавать клинический анализ крови. Больные средне-тяжелым и тяжелым НМС должны проходить лечение в отделении гематологии. Развитие гемолитического, апластического кризов из-за большого риска летального исхода являются показанием для перевода в отделение реанимации и интенсивной терапии.
Консервативная терапия
При уровне гемоглобина ниже 70 г/л производится переливание эритроцитарной массы, взвеси или отмытых эритроцитов. При гемоглобине меньше 50 г/л прибегают к переливанию цельной крови. Гемотрансфузию стоит выполнять медленно во избежание гемолитических реакций. При длительных гемотрансфузиях с целью выведения избытка железа обязательно используется хелатирующая терапия – дефероксамин, аскорбиновая кислота.
Для поддержания ремиссии (предотвращения кризов) больным тяжелым и средне-тяжелым НМС назначается постоянный прием профилактической дозы фолиевой кислоты. При апластическом кризе требуется дополнительное введение тромботического концентрата, стимуляторов лейкопоэза (филграстим), антибиотиков широкого спектра действия (цефтриаксон). Трофические язвы обрабатываются антисептическими растворами (фурацилин), мазями, содержащими антибиотики.
Хирургическое лечение
Основной радикальный вид лечения, обеспечивающий выздоровление больного, – тотальная спленэктомия (полное удаление селезенки). Она показана пациентам с частыми гемолитическими кризами, глубокой анемией, выраженной гипербилирубинемией и спленомегалией. Оптимальный возраст для операции – 6 лет. Предпочтение отдается лапароскопическому вмешательству как менее травматичному. Субтотальная резекция и эмболизация селезеночной артерии не рекомендуются, так как ассоциированы с высокой частотой рецидивов. При желчнокаменной болезни показана холецистэктомия.
Профилактика и прогноз
В целом наследственный микросфероцитоз является доброкачественным заболеванием. Подавляющее число пациентов имеет легкую или бессимптомную форму с незначительной спленомегалией и компенсированным гемолизом. Летальные исходы крайне редки (1-2%) и связаны с кризами (гемолитическими и апластическими). После спленэктомии продолжительность жизни не отличается от таковой у общей популяции. Первичная профилактика не разработана.
Отсутствие селезенки увеличивает риск инфицирования инкапсулированными микроорганизмами. Поэтому перед операцией (за 2-3 недели) обязательно проведение вакцинации против пневмококка, менингококка и гемофильной палочки. Дети до 6 лет должны получать профилактические дозы пенициллиновых антибиотиков (амоксициллин). Также с целью предотвращения постспленэктомического тромбоза назначаются антикоагулянты (низкомолекулярные гепарины).
Наследственный сфероцитоз (микросфероцитоз или болезнь Минковского-Шоффара) характеризуется гемолитической анемией внутриэритроцитного вида, ахолурической желтухой, высоким показателем ретикулоцитоза и спленомегалией.
К тому же отмечаются характерные эритроцитные признаки, в том числе микросфероцитоз, ослабление устойчивости эритроцита к гипотонному солевому раствору, повышение показателя самопроизвольного гемолиза (который выравнивается после назначения глюкозы), секвестрация эритроцитов в селезенке и положительный результат после удаления последней.
История изучения наследственного сфероцитоза - микросфероцитоза
Ценный вклад внес Шоффар, который, в 1907 г., определил наследственный характер гематологического расстройства, при котором развивается микроцитоз эритроцитов и понижение их устойчивости.
Географическое распространение и частота наследственного сфероцитоза - микросфероцитоза
Распространение болезни носит повсеместный характер — встречается на всех материках, среди всех рас и лиц обоего пола. Наблюдаются некоторое преобладание среди белой расы, в частности у северноевропейских народов и относительная редкость случаев заболевания среди негров Африки и Америки. У нас в стране представляется как гемолитическая болезнь внутриэритроцитного вида наибольшого распространения.
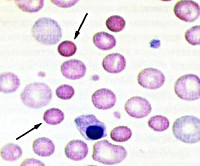
Мазок крови при наследственном сфероцитозе
Этиология наследственного сфероцитоза - микросфероцитоза
Болезнь передается преобладающим аутосомальным менделеевским способом (переходит по наследтсву от одного из родителей, независимо от пола, причем поражаемые лица гетерозиготные в отношении отягчений, болезнь выявляется у 50% потомства). Описаны и отдельные случаи, объяснение которых следует искать в мутациях, рождении незаконных детей или невысокой проницаемости отягчений у предшественников.
Наличие гомозиготных форм еще не доказано; видимо подобный продукт зачатия нежизнеспособный (Израель).
Клинические признаки наследственного сфероцитоза - микросфероцитоза
Начало наследственного сфероцитоза - микросфероцитоза. Несмотря на наследственный характер болезни лишь в редких случаях она выявляется вскоре после рождения или в раннем детстве. Чаще заболевание обнаруживается в условиях соматических преобразований на втором периоде детского возраста (7—8 лет) или у подростков. В отдельных случаях, при купании матерями обнаруживалось наличие отягчения уже на первый месяцах жизни ребенка, когда нашупывалась увеличенная селезенка (личные неопубликованные случаи).
Однако описаны и случаи проявления характерной картины заболевания лишь в возрасте 75 или даже 80 лет, что говорит о различном проникновении отягчения и хорошей компенсации на весьма продолжительный период времени. Конечно, болезнь выявима и раньше, но при условии проведения семейного исследования в попытке обнаружения скрытых случаев.
Формы проявления мало убедительны, в основном это признаки строения и общего характера (задержка физического развития с заниженной сосредоточенностью у детей школьного возраста, а у взрослых — неуспеваемость в работе коллектива; также описаны нехарактерные явления диспепсии). Вот почему период начала следует рассматривать больше как период отождествления наличия болезни.
Период разгара наследственного сфероцитоза - микросфероцитоза определяется по следующим характерным признакам:
а) Нарушения соматопсихического развития — отмечающиеся в большинстве случаев. У больных нередко coxpai яется детский аспект, в наступлении половой зрелости отмечается задержка (недоразвитие роста и веса и слабое половое развитие); при этом инфантилизм сопровождается и другими эндокринными растройствами ((Wintrobe).
б) Аспект покровов и слизистых оболочек. Бледность и желтушная окраска два элемента, которые, нередко, чередуются или проявляются одновременно. Тем не менее бледность покровов не чрезмерно резкая, поскольку костный мозг располагает большой способностью компенсации. Составляют исключение тяжелые анемии, при которых продолжительность жизни эритроцитов сокращается до менее 18—20 дней и процесс кроветворения не покрывает высокую степень разрушения красных кровяных клеток.
в) Спленомегалия. Кроме бледности и желтухи спленомегалия составляет третий основной признак заболевания. Ее размеры колеблятся от едва ли прощупываемой селезенки до занимающей всю левую часть брюшной полости. В принципе она не чувствительна при прощупывании и хотя может быть обнаружена уже на первом году жизни, она постепенно увеличивается, достигая максимальной величины у взрослого человека. Вопреки тому факту, что спленомегалия последствие гемолитической гиперактивности, не установлена какая-либо взаимосвязь ее размера и клинического состояния.
Гигантская спленомегалия обусловливает нарушения, связанные с компрессией, а иногда болезненный периспленит, способствующий образованию распространенной спайки с прилегающими зонами, (Heilmeyer L., Busch D.).
г) Хроническое изъязвление голеней отмечается примерно в 1/3 случаев. В основном оно локализуется в дистальной части, возможно одной или обеих голенях. Изъязвление длится месяцы и даже годы, после выздоровления остаются зоны гиперпигментации, которые перемежаются с перламутровыми участками. Травма, будь она и небольшая, способствует развитию изъязвления. Наличие этого трофического нарушения составляет ценное доказательство внутриэритроцитной гемолитической анемии.
Поздний период наследственного сфероцитоза - микросфероцитоза. Поскольку болезнь наследственного характера ее течение отличается чередованием периодов улучшения и обострения болезни. Существенным элементом для течения болезни представляется сохранение или нарушение равновесия между повышенным расплавлением крови и высоким показателем восстановления красных кровяных клеток. В результате этих колебаний анемия и желтуха исчезают или обостряются. Когда оба явления клинически исчезают, лишь спленомегалия свидетельствует о наличии болезни и трофические расстройства голеней, естественно когда в прошлом бывали изъязвления.
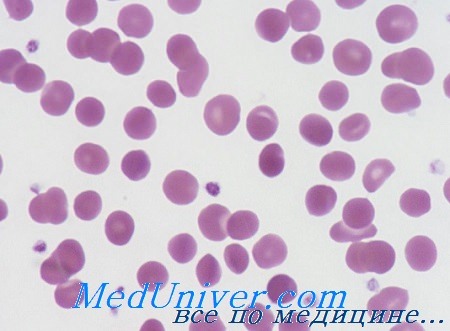
Мазок крови при наследственном сфероцитозе
Осложнения наследственного сфероцитоза гематологические или желчнопеченочные
Наступление приступа обусловливают весьма разнообразные причины, в том числе, инфекция, чрезмерное потребление спиртных напитков, физическое усилие, беременность, послеродовой период, менструация. В отдельных случаях диспептическое состояние, психическая травма или даже метеорологическое влияние благоприятствуют подобным приступам, а иногда и переливание совместимой крови. Однако нередко отсутствует какая-либо видимая причина, за счет которой можно отнести развитие приступа.
В принципе больные жалуются на общее плохое состояние, к которому относятся слабость (вплоть до прострации), сердцебиение, головная боль, тошнота, боль в животе и даже рвота, озноба и лихорадочное состояние (с повышением температуры до 39—40°С). Иногда наблюдается быстрое увеличение объема селезенки. В тяжелых случаях отмечаются коллапс и кома, требующих срочное определение в больницу, массивные переливания крови, за отсутствием которых может наступить летальный исход. Однако, помимо тяжелых форм, значительно чаще развиваются умеренные приступы, проявляющиеся усилением анемии и желтухи, которым сопутствуют слабость, явления диспепсии, гиперхромная моча и пр.
Длительность регенеративной фазы также колеблется. Так, при легких формах она составляет 3—4 дня, в то время как в тяжелых — 10—15 дней. С клинической точки зрения состояние больных возвращается к исходному — предприступному.
б) Желчно-печеночные осложнения наследственного сфероцитоза наблюдаются, в основном, у лиц в возрасте, отнюдь не составляя исключение даже у детей. У взрослых поражение желчного пузыря составляет 1/2 случаев, оно проявляется простым воспалением желчного пузыря, но наиболее часто желчнокаменным пигментным заболеванием (причем камни образованы из билирубината кальция). В некоторых случаях каменная болезнь осложняется резью в печени и явлениями ангиоколита.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
В статье приведен обзор литературы, касающийся клинических и лабораторных проявлений, диагностики, лечения и прогноза наследственной микросфероцитарной анемии. Описаны результаты собственных наблюдений двоих пациентов, находящихся на лечении в онкогематологическом отделении Йошкар-Олинской детской городской больницы.
Hereditary microspherocytosis (Minkowsky — Shauffard disease)
The paper presents a literature review of clinical and laboratory manifestations, diagnosis, treatment and prognosis of hereditary microspherocytic anemia. It is describes the results of his own observations of two patients treated in oncohematological office in Yoshkar-Ola City Children’s Hospital.
Наследственный микросфероцитоз был впервые описан в 1900 году Минковским, а в дальнейшем более подробно — Шоффаром. Распространенность данного заболевания — 1:5000 населения, наиболее часто встречается у жителей Северной Европы [1, 2].
Патогенез. Врожденная микросфероцитарная гемолитическая анемия представляет собой семейное заболевание, наследуемое по аутосомно-доминантному типу. В основе заболевания лежит генетический дефект белка мембраны эритроцитов, в результате чего повышается ее проницаемость для ионов натрия, что приводит к набуханию эритроцитов, нарушению способности эритроцитов деформироваться, отщеплению в селезенке части их поверхности, укорочению продолжительности их жизни и разрушению макрофагами селезенки [3, 4]. Патология эритроцитов проявляется морфологической аномалией — микросфероцитозом. Продолжительность пребывания микросфероцитов в циркулирующей крови резко уменьшена, средний срок их пребывания в кровеносном русле может составлять 12-14 дней (вместо нормальных 120-125 дней) [5].
Рисунок 1. Морфология микросфероцитов в мазках периферической крови больного (слева), здорового (справа)

Дифференциальная диагностика наследственного микросфероцитоза сводится к диагностике гемолитической анемии вообще. Многим больным ошибочно ставят диагноз синдрома Жильбера, хронического гепатита или даже цирроза печени, а анемию считают следствием этих заболеваний. Во всех случаях желтухи необходимо тщательное обследование больного независимо от содержания гемоглобина. Преимущественное увеличение содержания непрямого билирубина, ретикулоцитов, выявляемый при просмотре мазка микросфероцитоз дают основание для правильного диагноза. Затем необходима дифференциальная диагностика аутоиммунной гемолитической анемии и микросфероцитоза, так как аутоиммунные гемолитические анемии часто дают симптоматический микросфероцитоз [3]. В этой ситуации помогают тщательное выяснение анамнеза, давности заболевания, наличие подобного заболевания у родственников, выявление изменений скелета, постановка проб, позволяющих выявить аутоантитела (прямая проба Кумбса).
Лечение и прогноз. Лечение во время гемолитического криза направлено на ликвидацию анемии, гипоксии, гипербилирубинемии. Методом выбора при лечении наследственного сфероцитоза является спленэктомия, оптимальная в возрасте 4-5 лет. Спленэктомия обеспечивает практическое выздоровление, несмотря на сохранность сфероцитоза. Показанием к спленэктомии при микросфероцитозе являются постоянная или возникающая в виде кризов анемия, значительная гипербилирубинемия, даже без анемии, появление болей в правом подреберье, отставание в развитии у детей [3]. Переливания крови (эритроцитарной массы) показаны в случае тяжелого гемолитического криза [5]. Прогноз при наследственном микросфероцитозе благоприятный. Вероятность развития заболевания у детей, если один из супругов болен микросфероцитозом, несколько ниже 50% [9].
Клинический случай № 1. Больной С., 1 год 10 мес., с 01.06.09 г. по 09.06.09 г. находился на стационарном обследовании в онкогематологическом отделении Йошкар-Олинской детской городской больницы. Из анамнеза известно, что ребенок от первой беременности, родился недоношенным, с массой тела 900 грамм. Выхаживался в отделении второго этапа выхаживания недоношенных. Наследственность неизвестна, так как в четырехмесячном возрасте был усыновлен. С рождения отмечались снижение уровня гемоглобина в анализе крови, иктеричность кожных покровов. С декабря 2008 года наблюдается гематологом по поводу анемии недоношенных, принимал препараты железа. При поступлении жалобы на вялость, слабость, повышение температуры до фебрильных цифр, бледность и желтушное окрашивание кожных покровов. При пальпации печень выступает из-под края реберной дуги на 2,5 см, селезенка — на 2 см, мягкая, эластичная.
Общий анализ крови от 01.06.09 г.: HGB — 106 g/l, RBC — 3,9×10¹²/l, WBC — 11,6×10 9 /l, Ht — 26%, MCV — 68,8 fl, MCH — 25,8 pg, МСНС — 375 g/l, RDW — 20,5%, PLT — 434×10 9 /l, СОЭ — 6 мм/ч, в лейкоцитарной формуле: эозинофилы — 10%, с/я нейтрофилы — 34%, лимфоциты — 47%, моноциты — 9%.
Ребенок выписан в стабильном состоянии 09.06.09 г. с рекомендациями: наблюдение гематолога и педиатра, общий и биохимический анализы крови 1 раз в 3 месяца.
24.06.09 г. больной С. повторно поступает в онкогематологическое отделение. При поступлении состояние тяжелое, жалобы на вялость, отсутствие аппетита, плохой сон, повышение температуры до 39,6°С, бледность, желтушность кожных покровов, темный цвет мочи. Зев гиперемирован, рыхлый. Увеличены лимфатические узлы шейной группы. Живот увеличен в объеме, печень выступает из-под края реберной дуги на 3,5 см, селезенка — на 5 см.
Общий анализ крови от 24.06.09 г.: HGB — 89 g/l, RBC — 3,4×10¹²/l, WBC — 27,7×10 9 /l, Ht — 21%, MCV — 68,6 fl, MCH-25,5 pg, МСНС — 381 g/l, RDW — 20,8%, PLT — 260×10 9 /l, СОЭ — 12 мм/ч, в формуле: эозинофилы — 4%, базофилы — 1%, п/я нейтрофилы — 4%, с/я нейтрофилы — 15%, лимфоциты — 20%, моноциты — 1%, атипичные мононуклеары — 55%. Ретикулоцитоз — 46,7%. В мазках крови до 50% микросфероцитов. Непрямой билирубин — 41 мкмоль/л, АЛТ — 19 Е/л, АСТ — 45 Е/л, щел. фосфатаза — 316 Е/л, ЛДГ — 295 Е/л. Определяется повышенное содержание стеркобилина в кале. В моче отмечается уробилинурия, выделение уратов и кристаллов мочевой кислоты, объясняемое повышенным распадом эритроцитов. ИФА на вирус Эпштейна — Барр (инфекционный мононуклеоз) — положительный.
Учитывая у ребенка наличие интоксикации, лихорадки, увеличение шейных групп лимфоузлов, в крови высокий лейкоцитоз, мононуклеары до 55%, положительный ИФА, был диагностирован инфекционный мононуклеоз, который в свою очередь явился пусковым моментом для развития у мальчика гемолитического криза.
Проведено лечение: диета, постельный режим, инфузионная и противовирусная терапия. В результате терапии лихорадка купирована, уменьшились размеры печени, селезенки и лимфатических узлов. Общий анализ крови от 06.07.09 г.: HGB — 104g/l, RBC — 3,9×10¹²/l, WBC — 10,7×10 9 /l, Ht — 28%, MCV — 73,3 fl, MCH — 26,4 pg, МСНС — 360 g/l, RDW — 21,8%, PLT — 452×10 9 /l, СОЭ — 2 мм/ч, в лейкоцитарной формуле: эозинофилы — 5%, п/я нейтрофилы — 5%, с/я нейтрофилы — 25%, лимфоциты — 47%, моноциты — 8%, атипичные момонуклеары — 10%. Ретикулоцитоз — 9,3%. Количество микросфероцитов снизилось до 23%. Непрямой билирубин — 18,8 мкмоль/л. Нормализовались аппетит и сон. В стабильном состоянии 09.07.09 г. ребенок выписан домой с рекомендациями: наблюдение гематолога 1 раз в 3 месяца, общий анализ крови с ретикулоцитами и биохимический анализ крови 1 раз в 3 месяца, контроль УЗИ брюшной полости 1 раз в год.
Клинический случай № 2. Больная Г., 12 лет, 22.09.09 г. переведена в онкогематологическое отделение Йошкар-Олинской детской городской больницы из инфекционного стационара. При поступлении жалобы на заложенность носа, боль в горле, общую слабость, увеличение шейных и затылочных лимфоузлов, лихорадка до 39,2°С, иктеричность кожных покровов, гепатоспленомегалия с выраженной плотностью паренхиматозных органов. Из анамнеза известно, что отец девочки страдает синдромом Жильбера и у девочки с рождения на фоне повышения температуры, ОРВИ — появляется желтушность кожи, склер.
Общий анализ крови от 23.09.09 г.: HGB — 61g/l, RBC — 1,8×10¹²/l, WBC — 10,7×10 9 /l, Ht — 16%, MCV — 85 fl, MCH — 32,5 pg, МСНС — 382g/l, RDW — 20,3%, PLT — 162×10 9 /l, СОЭ — 48 мм/ч, в лейкоцитарной формуле: миелоциты — 1%, метамиелоциты — 2%, п/я нейтрофилы — 29%, с/я нейтрофилы — 9%, лимфоциты — 24%, моноциты — 4%, атипичные мононуклеары — 31%. Ретикулоцитоз — 82,5%. В мазках крови значительное количество микросфероцитов. Осмотическая резистентность эритроцитов: min — 0,72%, max — 0,42%, определяется резкое понижение резистентности по отношению к гипотоническим растворам поваренной соли. Повышено содержание уробилина в моче и стеркобилина в кале. Биохимический анализ крови от 23.09.09 г.: АЛТ — 71 Е/л, АСТ — 78 Е/л, ЛДГ 1234 Е/л, билирубин прямой — 5,68 мкмоль/л, непрямой — 54,8 мкмоль/л. Прямая проба Кумбса — отрицательная.
Тяжелая нормохромная анемия, обнаружение значительного количества микросфероцитов в препаратах крови, выраженный ретикулоцитарный криз, резкое понижение осмотической резистентности эритроцитов, гипербилирубинемия, а также наличие сходной симптоматики у отца дали основание диагностировать у девочки гемолитический криз наследственной микросфероцитарной анемии, спровоцированный инфекционным мононуклеозом (ИФА на вирус Эпштейна — Барр — положительный). Осмотр инфекциониста подтверждает диагноз инфекционного мононуклеоза.
Больной были назначены инфузионная, антибактериальная и противовирусная терапия, витамины. На фоне лечения состояние пациентки стабилизировалось. Общий анализ крови от 12.10.09 г. : HGB — 106g/l, RBC — 3,5×10¹²/l, WBC — 3,9×10 9 /l, Ht — 29,3%, MCV — 83,8 fl, MCH — 30,4 pg, МСНС — 362 g/l, RDW — 217,8%, PLT — 170×10 9 /l, СОЭ — 8 мм/ч, в формуле: базофилы — 1%, п/я нейтрофилы — 4%, с/я нейтрофилы — 62%, лимфоциты — 20%, моноциты — 4%, атипичные мононуклеары — 9%. Ретикулоцитоз — 13,1%. Биохимический анализ крови: АЛТ — 41 Е/л, АСТ — 32 Е/л, билирубин прямой — 2,75 мкмоль/л, непрямой — 24,35 мкмоль/л. По данным УЗИ брюшной полости отмечается положительная динамика в виде уменьшения размеров печени и селезенки. Девочка выписана домой 13.10.09 г. с рекомендациями: наблюдение гематолога, инфекциониста и педиатра, общий анализ крови с ретикулоцитами 1 раз в 3 месяца, контроль УЗИ брюшной полости.
Оба пациента находятся на диспансерном учете у врача-гематолога.
Диагноз врожденного микросфероцитоза представляет порой известные трудности. Наиболее типичные проявления гемолиза — желтушность склер и кожных покровов — не всегда бывают правильно расценены врачом. Описанные нами клинические случаи являются прямым подтверждением этого. У обоих пациентов с рождения определялись иктеричность склер и кожных покровов, но подробного обследования, до развития гемолитического криза, больным не проводилось. Наличие сходной симптоматики у отца девочки (синдром Жильбера?) дает возможность предположить, что пациентка Г. заболевание унаследовала от отца. Наследственность больного С. неизвестна.
В обоих случаях провоцирующим фактором развития гемолитического криза явился вирус Эпштейна — Барр. Дети и раньше болели вирусными инфекциями, но развития гемолитического криза они не вызывали. Возможно, не всякая, а лишь определенная группа вирусов может вызывать кризы. Этот вопрос остается открытым, так как на примере двух случаев нельзя утверждать, что вирус Эпштейна — Барр всегда провоцирует развитие гемолитического криза у больных наследственным микросфероцитозом.
Е.В. Ширданина, З.С. Гордеева
Йошкар-Олинская детская городская больница, г. Йошкар-Ола
Ширданина Екатерина Валерьевна — врач клинико-диагностической лаборатории
Читайте также: