
Биохимические скрининг тесты для выявления наследственных ферментопатий реферат
Обновлено: 08.07.2024
Частота тяжёлых наследственных и врождённых заболеваний в последнее время немного снизилась, благодаря достижениям в генетике. Успеху способствовало активное внедрение методов, с помощью которых патологии развития плода выявлялись ещё на стадии беременности. В случае выявления тяжёлых нарушений в развитии плода, беременность рекомендуется прервать. По этой причине больных детей сейчас рождается меньше, но частота выявления наследственных заболеваний остаётся на прежнем уровне.
Наиболее часто встречается такая наследственная патология, как синдром Дауна (трисомия по хромосоме 21). Своевременная профилактика этого заболевания целесообразна не только с медицинской точки зрения. Сокращения количества детей, страдающих данной патологией, имеет психологическое, социальное, и экономическое значение.
Особенности профилактики
Для того, чтобы на ранней стадии беременности выявить наличие у плода синдрома Дауна, дефекта невральной трубки (ДНТ) и синдрома Эдвардса, имеются все условия и возможности. Диагностика осуществляется двумя способами:
- проведение исследований индивидуально у каждой женщины, вошедшей в группу риска, при помощи молекулярно-генетических и цитогенетических и технологий, которые предоставляют возможность исследовать клетки плода на молекулярном и хромосомном уровнях.
Технологии диагностики патологии плода в 1-ом и 2-ом триместре беременности по биохимическим маркерам в последнее время активно внедряются в практику. Проводятся два вида тестов – двойной тест (определение свободного бета-ХГЧ и ПАПП-А-белка), и тройной тест (определение ХГЧ, АФП, НЭ с последующим расчётом риска возникновения патологии).
Двойной тест
Хорионический гонадотропин - свободная субъединица бета-ХГЧ – это оптимальный маркер, позволяющий выявить патологию развития плода в 1-м триместре беременности. Клетки трофобласта начинают осуществлять синтез ХГЧ после того, как эмбрион будет имплантирован в полость матки. Этот процесс продолжается всю беременность. Наибольшая концентрация ХГЧ приходится на 11-12-ю неделю беременности, затем его количество начинает снижаться.
ПАПП-А-белок продуцирует плацента, причём его количество резко возрастает с наступлением беременности. Это лучший гормональный маркер 1-ого триместра, который позволяет достоверно диагностировать наличие у плода синдрома Дауна. Тестирование беременных женщин осуществляется, как правило, при сроке 9-14 недель.
Тройной тест
Гормоны АФП, ХГЧ и свободный эстриол входят в число основных маркеров, которые позволяют определить патологии развития плода во 2-м триместре беременности.
АФП (альфа-фетопротеин) представляет собой вещество, вырабатываемое печенью плода. В кровь женщины это вещество попадает через плаценту. Если у плода существуют такие патологии развития, как незаращение передней брюшной стенки, спинномозговые или черепно-мозговые или грыжи и т.д., то концентрация АФП в крови женщины резко повышается. Снижение концентрации АФП отмечается при наличии у плода синдрома Дауна.
Определения в крови беременной женщины уровня НЭ (неконъюгированный (несвязанный, свободный) эстриол) и ХГЧ (хорионический гонадотропин) также помогает диагностировать синдром Дауна. Такие тестирования осуществляются при сроке беременности с 14-ой по 21-ю неделю.
Для всех вышеперечисленных исследований используется сыворотка крови женщины. Анализы проводят на голодный желудок, как и все другие гормональные исследования. Как правило, результаты анализов выдают на 3-4-й день или через несколько часов в случае срочности.
Теория вероятности
Скрининговое исследование позволяет определить риск возникновения у плода различных патологий. Лучше всего проводить исследование с использование специальной компьютерной программы, которая принимает во внимание индивидуальные особенности женщины.
Отклонения от нормальных показателей биохимических маркеров могут быть спровоцированы несколькими факторами:
- наличие в анамнезе акушерской патологии;
- вид оплодотворения, например, ЭКО;
- воздействие лекарственных (гормональных) препаратов;
Все эти факторы учитываются программой. На руки пациентка получает бланк ответа, который включает в себя все данные, которые могут потребоваться врачу-клиницисту. Для того, чтобы определить, насколько велик риск развития наследственной или врожденной патологии, врачу необходимо обработать все данные. Но поставить диагноз лишь на основании этих анализов невозможно. Для того, чтобы подтвердить или исключить возможные предположения, пациентке назначают дополнительные методы обследования, с использованием амниоцентеза и кордоцентеза - цитогенетических исследований клеток плода.
Если анализ показал, что биохимические маркеры имеют показатели выше или ниже нормальных значений – это повод для беспокойства. Но беременной женщине не следует впадать в панику. Врачи контролируют ситуацию и сумеют предотвратить негативные последствия.

БИОХИМИЧЕСКИЕ МЕТОДЫ ДИАГНОСТИКИ МОНОГЕННЫХ ПАТОЛОГИЙ
Текст работы размещён без изображений и формул.
Полная версия работы доступна во вкладке "Файлы работы" в формате PDF
Моногенные болезни — разнородная по клиническим проявлениям группа заболеваний, обусловленных мутациями на уровне гена. [3, с. 118]
В основу классификации моногенных болезней положено несколько принципов, но с клинической и биохимической точки зрения наиболее важным является принцип классификации - по преимущественному поражению того или иного вида обмена. Благодаря этому многие МБ называются наследственными болезнями обмена веществ (НБО). Среди них выделено более 700 форм, в том числе 200 с установленным биохимическим дефектом.
Среди НБО выделяют:
1.болезни аминокислотного обмена (ФКУ, тирозиноз, алкаптонурия, лейциноз и др.);
2. болезни углеводного обмена (галактоземия, гликогенозы, мукополисахаридозы);
3.болезни липидного обмена (эссенциальные семейные липидозы, ганглиозидозы, сфинголипидозы, цереброзидозы, лейкодистрофии, гиперлипидемии и др.);
4.болезни биосинтеза кортикостероидов (адрено-генитальный синдром, гипоальдостеронизм и др.);
5. болезни пуринового и пиримидинового обмена (оротоваяацидурия, подагра и др.);
6. болезни порфиринового и билирубинового обмена (синдромы Жильбера, Криглера-Найяра, порфирии и др.);
7. болезни эритрона (анемия Фанкони, гемолитические анемии, дефицит глюкозо-6-фосфатдегидрогеназы и др.);
8. болезни металлов (болезни Вильсона-Коновалова, Менкеса, семейный пери-одический паралич и др.);
9. болезни транспорта систем почек (болезнь де Тони-Дебре-Фанкони, витамин D-резистентный рахит, тубулопатии и др.);
10. болезни лимфоцитов и лейкоцитов (недостаточность аденозиндезаминазы, септический гранулематоз и др.). [1, с.209]
До недавнего времени диагностика моногенных наследственных болезней основывалась исключительно на особенностях фенотипического проявления заболевания. Только при некоторых наследственных болезнях обмена веществ диагностику проводили, определяя уровень измененных метаболитов или даже измененного фермента. Фенотипический уровень диагностики иногда был достаточным для решения некоторых проблем клинической генетики.[2, с.132]
В настоящее время существует широкий спектр методов для диагностики моногенных патологий.
Могут использоваться следующие методы:
1) Портретная диагностика – для диагностики моногенных синдромов, проявляющихся пороками развития.
2) Биохимические методы – для диагностики наследственных нарушений обмена веществ.
3) ДНК- диагностика для ферментопатий и др.
4). Цитогенетические методы – для диагностики синдрома фрагильной Х-хромосомы. [3, с.121]
Разнообразные биохимические методы используют для диагностики моногенных болезней с выявленным биохимическим дефектом из группы наследственных болезней обмена. Биохимическая диагностика имеет тем большую ценность, чем меньше клинических проявлений в ранних стадиях болезни. чем надежнее диагностические возможности гетерозиготного носительства, чем меньше стоимость этих исследований по сравнению с молекулярно-генетическими, иммуногенетическими и др.
Биохимические методы являются уникальными при массовом скрининге для ранней диагностики наследственных болезней у новорожденных. [2, с.135]
Критериями для включения патологии в программу массового биохимического скрининга новорожденных являются следующие:
• высокая частота встречаемости в популяции (не менее 1:20 000);
• тяжесть поражения с необратимыми последствиями в виде инвалидизации или ранней смерти;
• возможность простой, надежной (чувствительной, специфичной, без ложноотрицательных результатов) и экономически оправданной диагностики в доклинической стадии заболевания;
• возможность патогенетической или иной эффективной коррекции выявленного нарушения. [3, с.122]
Для целей массового скрининга чаще всего используют капиллярную кровь, которую берут из пятки новорожденного на 3-4-й день жизни и пропитывают ею с двух сторон отпечатанные на фильтровальной бумаге (специальные тест-полоски) кружочки в количестве, соответствующем числу диагностируемой патологии. Образец с высушенной кровью отправляется по почте в специализированные генетические диагностические центры, где обеспечивается экспрессдиагностика. При выписке из роддома мать получает на руки документ, в котором, наряду с данными о течении родов и параметрах новорожденного, указывается информация о том, на какие НБО у ребенка взяли анализы и дата забора биологического материала (крови). Семью извещают о результатах обследования только при выявлении патологии для проведения второго этапа - уточняющей диагностики и скорейшего назначения лечения. О результатах без патологий не сообщается. В условиях массового скрининга ребенок может быть не обследован в случае планированных домашних родов, при тяжелой перинатальной патологии, требующей интенсивной терапии или хирургического вмешательства.
К генетическим биохимическим методам диагностики относят:
• количественные. [3, с.123]
Кроме крови, ее плазмы, сыворотки и форменных элементов, для биохимических исследований могут использоваться моча, пот, культуры клеток (фибробластов, лимфоцитов).
Качественные тесты дешевы, просты, чувствительны, позволяют выявить избыточные концентрации субстратов или их производных при ферментных блоках реакций, в которых они участвуют. Для качественных тестов обычно используют мочу. Качественные реакции делятся на универсальные, определяющие группу заболеваний с ведущим биохимическим дефектом (например, ЦПХ-тест при мукополисахаридозах, проба Бенедикта на редуцирующие вещества и др.), и специфические (тест на гомогентизиновую кислоту при алкаптонурии, тест на медь при болезни Вильсона-Коновалова и др.). [2, с.156]
Полуколичественные и количественные методы биохимической диагностики проводятся и с мочой, и с кровью. С их помощью можно разделить метаболиты, принадлежащие к одному классу химических веществ, и определить концентрации определенного вещества. К этим методам относятся бумажная, тонкослойная (одно- и двумерная) и другие виды хроматографии, электрофорез, хроматомассспектрометрия, спектрофотометрия, флуориметрия, высокоэффективная жидкостная хроматография, тандемная масс-спектрометрия (позволяет количественно определить до 3000 метаболических маркеров). Эти методы сложные, но высокоточные и требуют использования дорогостоящего оборудования. [2, с.159]
В настоящее время наблюдается высокая частота встречаемости моногенных патологий в популяции (не менее 1:20 000), наиболее часто встречаемыми заболеваниями являются : Фенилкетонурия, Врожденный гипотиреоз, Адреногенитальный синдром, Галактоземия, Муковисцидоз, Нейрофиброматоз, Миотоническая дистрофия. Они характеризуются высокой тяжестью поражения с необратимыми последствиями в виде инвалидизации или ранней смерти. Поэтому важнейшая задача на сегодняшний день- это точная диагностика заболеваний на ранних этапах их развития.
Список, используемой литературы
2.Медицинская генетика - Гинтер Е.К. 2003г.стр. 132 -198.
В ряде случаев (отсутствие или частичная информативность) ДНК-диагностика некоторых заболеваний может быть дополнена другими диагностическими исследованиями. В случае гемофилии А возможно прямое определение уровня фактора VIII свертывания крови в пуповинной крови плода после 20 нед беременности. ДНК-диагностика адреногенитального синдрома может бьпъ дополнена прямым исследованием содержания 17-ОН прогестерона в амниотической жидкости (АЖ). ПД синдрома ломкой Х-хромосомы нередко дополняют прямым цитогенетическим анализом культуры лимфоцитов пуповинной крови плода, ДНК-диагностика миодистрофии Дюшенна принципиально может быть дополнена иммуноцитохимическим анализом биоптатов скелетных мышц плода.
В случае муковисцидоза дополнительная информация о состоянии плода может быть получена при биохимическом исследовании активности ферментов АЖ в 17-19 нед беременности. Разработанный и широко применяемый в нашей лаборатории алгоритм ПД муковисцидоза приведен на рисунке.
Биохимические методы в пренатальной диагностике
Наиболее часто материалом для биохимических исследований в ПД является АЖ, получаемая путем амниоцентеза. АЖ образуется за счет секреторной активности клеток амниона на ранних эмбриональных стадиях развития и за счет первичной мочи плода — в более поздние сроки. Количественный и качественный состав околоплодных вод регулируется компонентами системы амнион - мать - плод; нарушение любого из них приводит к избытку или недостатку вод, влияет на биохимический и клеточный состав АЖ.
Особенности биохимического и клеточного состава АЖ на разных сроках беременности подробно рассмотрены в специальных сводках и обзорах.
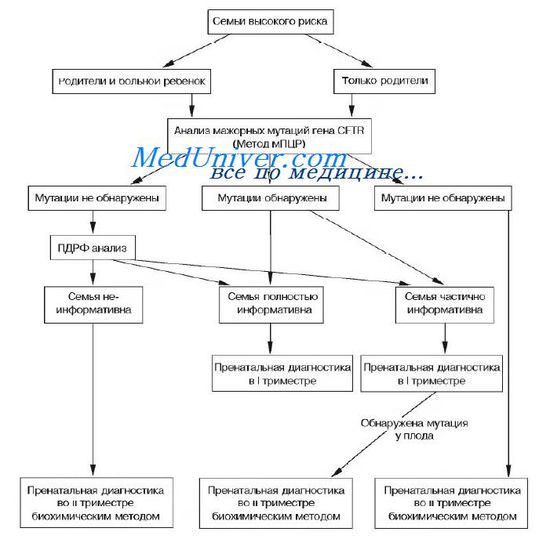
Принципиальная схема пренатальной диагностики муковисцидоза
Клетки амниотическои жидкости в биохимическом методе
Клетки АЖ используются в ПД не только для цитогенетического анализа, но и для выявления некоторых метаболических болезней, связанных с дефектами обмена веществ. Некультивированные клетки АЖ могут быть использованы для ПД некоторых наследственных дефектов обмена (НДО), когда с помощью гистохимических и электронно-микроскопических методов определяют специфические включения, характерные для ряда болезней накопления. Подобные способы ПД описаны для гликогеноза типа II, До появления ДНК-диагностики культура клеток АЖ долгое время была основным объектом ПД НДО.
Главное условие успешной ПД НДО — точная информация о первичном биохимическом дефекте, лежащем в основе заболевания. Большинство НДО наследуется по аутосомно-рецессивному типу, при котором оба родителя являются гетерозиготными носителями мутантного гена и с вероятностью 25% могут иметь больных детей.
Биохимический скрининг на гетерозиготное носительство для большинства НДО невозможен в связи с отсутствием средних значений концентраций исследуемых белков в норме. Молекулярный скрининг неоправдан экономически, за исключением ряда небольших этнических групп, в которых частота определенных мутаций существенно выше, чем в основной популяции. Примером могут служить ганглиозидоз GM2 типа IV у евреев-ашке-нази, лизосомные болезни типа сиалидо-за и галактосиалидоза, которые чаще встречаются у японцев и итальянцев.
Для точной ПД НДО недостаточно знания клинического диагноза. Даже при хорошо изученных симптомах болезни, например при ряде гликолипидозов и мукополисахаридозов, существуют варианты, обусловленные различными первичными биохимическими дефектами. Примером выраженной биохимической гетерогенности является гликогеноз типа I. Тип I Авызван дефектом глюкозо-6-фосфатазы, в то время как при типах В и С этого заболевания нарушения функционирования фермента обусловлены отсутствием других белков, необходимых для нормального метаболизма глюкозо-6-фосфатазы, а именно специфических транслоказ. При этом активность изучаемого фермента у одного из родителей может быть либо существенно выше, либо ниже среднего уровня и практически не отличаться от уровня, определенного у самого больного. Такая псевдонедостаточность описана, в частности для глобоидной лейкодистрофии.
Подобная вариабельность существенно снижает эффективность ПД биохимическими методами. Для успешной ПД в таких случаях особенно важна информация об экспрессии дефектного белка или фермента в разных тканях. Эта информация и определяет выбор оптимального биологического объекта ПД. Первичный биохимический дефект изучался в разных тканях только для некоторых групп ферментов (например, для лизосомных гидролаз). Кроме того, известно, что активность многих ферментов может варьировать в клетках разных тканей в пре- и постнатальном периодах.
Методы установления биохимического дефекта обычно связаны либо с оценкой активности фермента, либо с выявлением других белков, участвующих в ферментативной реакции, либо с определением продуктов, накапливающихся в результате ферментативного блока. В ряде специальных изданий можно найти подробное описание методов диагностики как давно известных, так и сравнительно недавно разработанных.
Основные наследственные болезни, для которых возможна ПД на основании биохимического исследования клеток АЖ или состава АЖ, приведены в таблице.
Каждой из перечисленных нозологических форм заболеваний соответствует свой первичный биохимический дефект, т.е. нарушение того или иного звена метаболизма, выяснение которого и составляет основную задачу биохимической диагностики.
В связи с быстрым накоплением знаний о геноме человека, идентификацией новых генов, исследованиями их мутаций и полиморфизмов, с одной стороны, сложностью и зачастую неопределенностью биохимических результатов, труднодоступностью специфических субстратов — с другой, диагностика все большего числа наследственных дефектов обмена проводится с использованием более универсальных и точных молекулярных методов анализа. Список НДО, для которых уже разработаны молекулярные методы диагностики, приведен в монографии В.Н. Горбуновой и B.C. Баранова.
Тем не менее некоторые белки АЖ, прежде всего так называемые эмбриоспецифические белки, т.е. белки, характерные только для плода и в норме не синтезирующиеся в организме матери, сохраняют большое диагностическое значение. К таким белкам в первую очередь следует отнести альфафетопротеин (АФП), ацетилхолинэстеразу, белки микроворсинок кишечника плода и стероидные гормоны.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.

Разработка программы скрининга началась в шестидесятых годах прошлого века, когда Роберт Гатри создал технологию тестирования сухих отпечатков крови на фильтровальной бумаге. Первым заболеванием, которое стало кандидатом для массовой диагностики, была фенилкетонурия, так как ее раннее выявление и коррекция питания способны предотвратить развитие тяжелых неврологических нарушений. Затем к скринингу добавилось еще несколько заболеваний: врожденный гипотиреоз, ВДКН, галактоземия и муковисцидоз. Тандемная масс-спектрометрия (ТМС) позволила значительно расширить список заболеваний, добавив к болезням обмена веществ гемоглобинопатии, спинальную мышечную атрофию, тяжелый комбинированный иммунодефицит и др.
Таблица 1 | Рекомендуемая The American College of Obstetricians and Gynecologists (ACOG) скрининг-панель для врожденных заболеваний
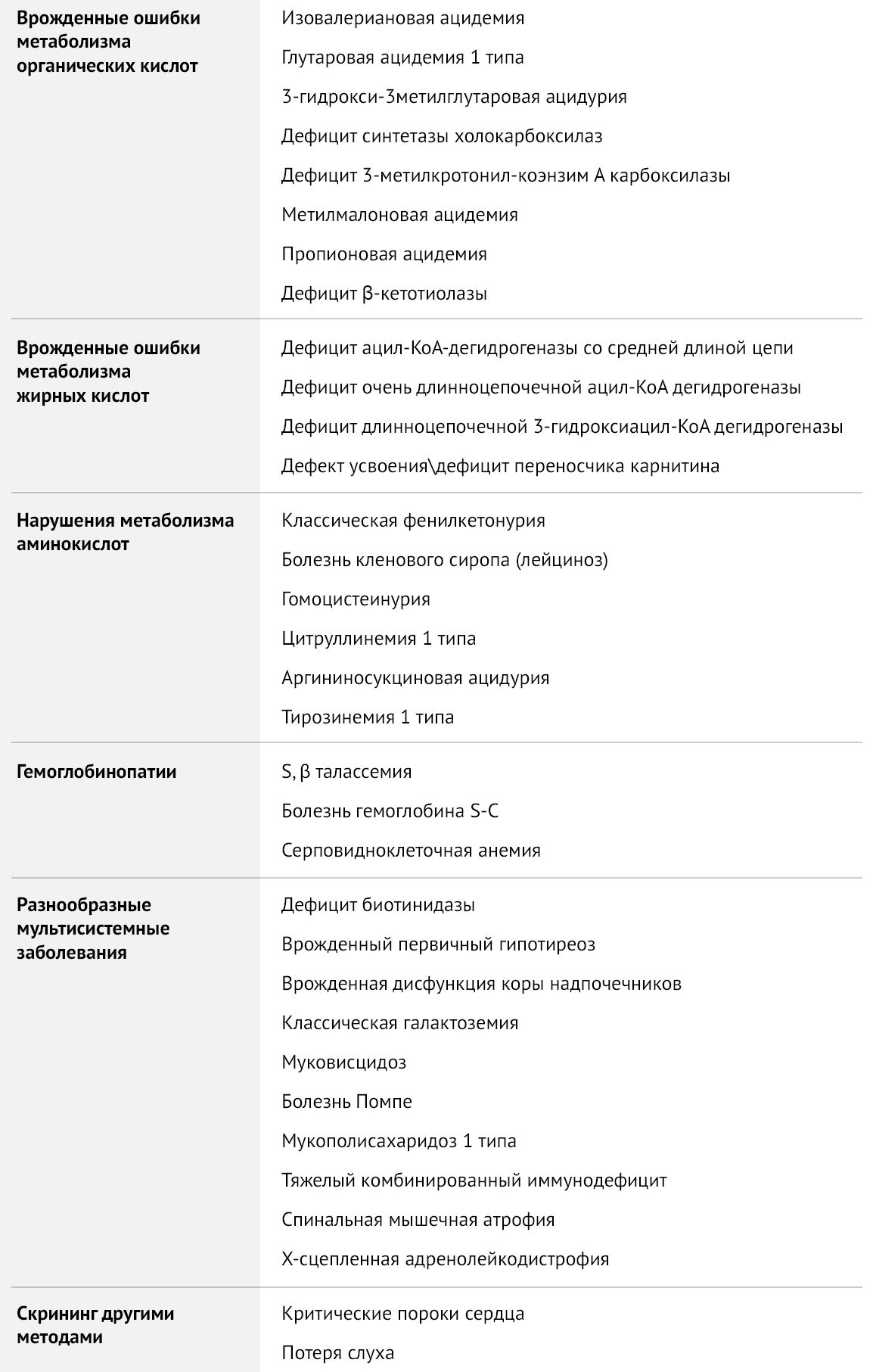
В данной статье будут рассмотрены два скрининга, доступные в нашей стране: обязательный, включающий тестирование на пять заболеваний (врожденный гипотиреоз, ВДКН, фенилкетонурия, галактоземия, муковисцидоз) и расширенный скрининг на наследственные нарушения метаболизма.
Фенилкетонурия (в современной классификации ― ФАГ-зависимая ФКУ) обусловлена мутацией гена фенилаланингидроксилазы и относится к числу аминокислотных аминоацидопатий. В норме фенилаланин (ФА) путем реакций гидроксилирования превращается в тирозин, однако в случае мутации вышеназванного гена активность превращающего фермента снижается, создается дефицит тирозина одновременно с избытком ФА, образующего токсичные метаболиты (фенилацетат, фенилпируват, фениллактат). Снижение образования тирозина влечет за собой нарушение синтеза гормонов щитовидной железы, нейротрансмиттеров и пигментов меланоцитов, а избыток ФА приводит к дисбалансу аминокислот в тканях мозга, обусловленному торможением их всасывания в желудочно-кишечном тракте или нарушением реабсорбции из почечных канальцев, нарушению образования или стабилизации полирибосом, снижению синтеза миелина, норадреналина и серотонина. Также за счет конкурентного ингибирования создается дефицит тирозиназы, что в совокупности с дефицитом тирозина приводит к снижению образования меланина и гипопигментации.
Основной проблемой пациентов с ФКУ являются нарушения функции ЦНС: от сонливости, вялости, отсутствия аппетита в период манифестации в 2–6 месяцев до тяжелых нарушений психомоторного развития в будущем; нередко развиваются атаксия, гиперкинезы, тремор рук, парезы по центральному типу. Единственный способ предотвратить развитие вышеназванных нарушений — назначение гипофенилаланиновой диеты с момента рождения с поддержанием низкого уровня фенилаланина в течение всей жизни.

Рисунок 1 | Интерпретация результатов исследования на наличие фенилкетонурии
ВДКН обусловлена дефицитом ферментов и транспортных белков, участвующих в биосинтезе кортизола. Наиболее часто встречается дефицит 21-гидроксилазы, что в свою очередь приводит к дефициту кортизола и альдостерона и ответному увеличению секреции АКТГ и гиперплазии коры надпочечников. В условиях дефицита фермента происходит значительное накопление предшественников гормонов, что приводит к увеличению синтеза тестостерона, не зависящего от 21-гидроксилазы. В итоге у пациента формируется надпочечниковая недостаточность и гиперандрогения. Гормональным маркером дефицита 21-гидроксилазы является уровень 17-гидроксипрогестерона (17-ОНП), определяемый в рамках неонатального скрининга. Результат трактуется как положительный, если при двукратном тестировании образца уровень 17-ОНП у доношенных новорожденных составляет ≥ 20 нг/мл. У недоношенных детей при заборе крови на 7–8 сутки после рождения скрининговый результат трактуется как положительный при следующих уровнях 17-ОНП: на сроке 23–32 недели гестации ― ≥ 65 нг/мл; на сроке 33–36 недель гестации ― ≥ 40 нг/мл.
Врожденный гипотиреоз в большинстве случаев вызван дефектами самой щитовидной железы (первичный гипотиреоз). Причины первичного врожденного гипотиреоза можно в широком смысле классифицировать как неспособность щитовидной железы нормально развиваться (дисгенезия) или неспособность структурно нормальной щитовидной железы производить нормальные количества гормона (дисгормоногенез). Дисгенезия щитовидной железы, охватывающая весь спектр агенеза, гипоплазии и эктопии, является наиболее частой причиной врожденного гипотиреоза. В то время как это заболевание остается наиболее частой причиной врожденного гипотиреоза, частота возникновения дисгормоногенеза за последние несколько десятилетий увеличилась. В то время как на дисгормоногенез приходится только 15 % врожденного гипотиреоза, диагностированного в первые дни скрининга новорожденных, у 30–40 % младенцев, прошедших скрининг по современным протоколам, имеется эктопическая щитовидная железа, соответствующая одной из форм дисгормоногенеза. В отличие от дисгенезии щитовидной железы, при которой моногенная причина присутствует только у небольшого количества пациентов, дисгормоногенез часто возникает из-за генетического дефекта на каком-либо этапе синтеза тиреоидных гормонов.
Учитывая разнообразие функций тиреоидных гормонов в организме человека, врожденный гипотиреоз характеризуется разнообразием клинических проявлений с поражением всех органов и систем. При отсутствии своевременного лечения на первый план выходит задержка психомоторного и речевого развития, затем наступают отставание в физическом развитии и задержка полового развития. Основной задачей скрининга является наиболее раннее выявление детей с подозрением на врожденный гипотиреоз.

Рисунок 2 | Интерпретация результатов исследования на наличие врожденного гипотиреоза
Галактоземия — аутосомно-рецессивное наследственное нарушение обмена углеводов, при котором в организме накапливается избыток галактозы и ее метаболитов. В норме галактоза образуется в результате гидролиза лактозы в кишечнике либо в процессе ферментных реакций, обмена гликопротеинов и гликолипидов. Галактоза является материалом для образования клеточных мембран, нервной ткани, нервных окончаний и т. д. В результате ферментных реакций она превращается в глюкозу, и именно дефицит галактозо-1-фосфатуридилтрансферазы лежит в основе патогенеза данного заболевания. Метаболиты галактозы обладают повреждающим действием. Так, галактитол проникает в хрусталик глаза, приводя к повышению осмотического давления, электролитным нарушениям и денатурации белка с формированием катаракты. Другие метаболиты обладают гепато-, нейро- и нефротоксическим действиями, а также вызывают гемолиз эритроцитов. Тормозящее влияние метаболитов галактозы на углеводный обмен приводит к гипогликемии.

Рисунок 3 | Интерпретация результатов исследования на наличие галактоземии
Муковисцидоз — аутосомно-рецессивное заболевание, связанное с мутацией гена МВТР (трансмембранного регулятора муковисцидоза). МВТР является хлорным каналом, мутации гена которого нарушают не только транспорт, но и секрецию ионов хлора. При затруднении их прохождения через клеточную мембрану увеличивается реабсорбция натрия железистыми клетками, нарушается электрический потенциал просвета, что вызывает изменение электролитного состава и дегидратацию секрета желез внешней секреции. В результате выделяемый секрет становится чрезмерно густым и вязким. Поражаются все экзокринные железы организма: печень, поджелудочная железа, мочеполовая система, но наиболее ярко муковисцидоз проявляет себя со стороны органов дыхания, провоцируя бронхообструкцию, дыхательную и сердечную недостаточность, легочную гипертензию.

Рисунок 4 | Интерпретация результатов исследования на наличие муковисцидоза
Органические ацидемии — группа аутосомно-рецессивных наследственных заболеваний обмена, в основе патогенеза которых лежит дефицит ферментов, участвующих в метаболизме белков, что приводит к повышению уровня кетоновых тел, обладающих токсическим действием на различные органы и ткани, в частности, на ЦНС. Данные заболевания манифестируют уже в стадии декомпенсации, как правило, в период с первой недели до первого года жизни. Триггерами служат стресс, длительное голодание, инфекционные заболевания, иммунизация, реже — чрезмерное употребление белковой пищи. Проявляются преимущественно неврологической симптоматикой: нарушение сознания вплоть до комы, эпилептические приступы, нарушение мышечного тонуса, у детей старшего возраста — нарушения психоречевого развития, атаксия, очаговые неврологические симптомы, синдром Рейе (острая печеночная недостаточность, сочетающаяся с энцефалопатией), психические расстройства.
Нарушения окисления жирных кислот — врожденный дефект метаболизма из-за нарушения либо митохондриального β-окисления, либо транспорта жирных кислот с использованием карнитинового транспортного пути. Проявления зависят от нарушения метаболизма конкретной кислоты, но все они имеют общие черты и требуют схожей тактики лечения. В периоде новорожденности метаболические нарушения проявляются тяжелой кардиомиопатией, гипокетотической гипогликемией, дисфункцией печени в первые несколько дней или недель жизни, часто заканчиваясь летально. В младенческом и детском возрасте характерны эпизоды летаргии и рвоты, развивается дисфункция печени и гипокетотическая гипогликемия, энцефалопатия, что может привести к внезапной младенческой смерти. У подростков и во взрослом возрасте дебютируют эпизодическим рабдомиолизом, мышечной слабостью, миалгией. Лечение включает отказ от голодания, симптоматическую терапию развившихся осложнений и включение в рацион добавок, если это необходимо.
Аминоацидопатии
Болезнь кленового сиропа (она же лейциноз) — наследственное заболевание, обусловленное дефицитом дегидрогеназы кетокислот с разветвленной цепью и нарушением метаболизма лейцина, изолейцина, валина (аминокислоты с разветвленной цепью, АКЦР). Повышение уровня АКЦР и их метаболитов, в частности, кетокислот, приводит к кетоацидозу, атрофии ткани головного мозга, нарушению окислительного фосфорилирования в дыхательной цепи митохондрий. Избыток лейцина обладает нейротоксическим эффектом, вызывая дисфункцию астроцитов, апоптоз нейронов и блокируя транспорт через гематоэнцефалический барьер аминокислот, важных для синтеза нейротрансмиттеров.
Гомоцистеинурия — наследственное заболевание из группы аминоацидопатий, обусловленное нарушением метаболизма серосодержащих аминокислот, в частности, метионина. Дефицит цистатион-b-синтазы нарушает преобразование метионина в цистеин. Высокий уровень гомоцистеина связан с образованием некротически-дегенеративных участков в почках, селезенке, слизистой оболочке желудка и сосудах, активацией XII фактора свертывания, способствующего тромбообразованию.
Аргининосукциновая ацидурия вызывается мутациями в гене ASL, который кодирует фермент аргининосукцинатлиазу. Этот фермент катализирует превращение аргинино-янтарной кислоты в аргинин и фумарат на четвертом этапе цикла мочевины. Дефекты на этой стадии цикла мочевины приводят к накоплению в плазме аммиака, аргинино-янтарной кислоты, цитруллина и оротовой кислоты в моче, а также к дефициту аргинина в плазме. Ацидурия может иметь различную клиническую картину с началом в любом возрасте, включая период новорожденности. Состояние новорожденных обычно не вызывает подозрений в течение первых 24–48 часов после рождения, но в течение нескольких дней дебютирует тяжелая гипераммониемия, проявляющаяся летаргией, сонливостью, отказом от еды, рвотой, тахипноэ и респираторным алкалозом. Если не начать лечение, может произойти обострение летаргии, судороги, кома и смерть. Позднее начало ацидурии обычно индуцировано острой инфекцией, стрессом или высоким потреблением белка. Сообщалось также о поздних когнитивных дефектах или нарушениях обучаемости при отсутствии эпизодов гипераммониемии. У некоторых пациентов заболевание может протекать бессимптомно, несмотря на четкие биохимические признаки.
Тирозинемия 1 типа — заболевание, обусловленное дефицитом фумарилацетоацетатгидролазы, в результате чего происходит накопление высокотоксичных фумарил- и малеилацетоацетата, обладающих гепатотоксическим и канцерогенным действием. Конечные метаболиты — сукцинилацетон и сукцинилацетоацетат — являются митохондриальными токсинами, тормозящими фосфорилирование и блокирующими цикл Кребса. Накопление токсинов приводит к прогрессирующему заболеванию печени с развитием печеночной недостаточности, цирроза, тубулопатии с формированием ренальной тубулопатии, гипофосфатемического рахита, синдрома Фанкони. Острая тирозинемия сопровождается развитием гипертрофической кардиомиопатии. Кроме того, нарушается путь синтеза порфирина, ингибируется синтез порфобилиногена, что приводит к кризам, проявление которых напоминает порфирию. Все пациенты подвержены высокому риску развития гепатоцеллюлярной карциномы, вторичной по отношению к циррозу. Без своевременного лечения дети погибают в возрасте 10 лет.
Неонатальный скрининг: как проводится?
Обследование на генетические заболевания проводят детям в первые 10 дней после рождения. Забор капиллярной крови из пяточки выполняют медицинские сотрудники родильного дома или поликлиники доношенным детям на 4 сутки жизни, а недоношенным — на 7 сутки жизни.
Почему скрининг проводят так рано? Такие сроки обследования связаны с тем, что болезни, на которые проверяют малышей, дебютируют в первые месяцы жизни и угрожают развитием тяжелых необратимых осложнений.
Лечение, начатое до появления первых симптомов, увеличивает шансы на успех. Ребенок сможет полноценно расти и развиваться наравне со сверстниками.
Пребывая в хлопотах и заботах, родители порой забывают о том, что их малышу проводили скрининг. Действительно, ведь при отсутствии отклонений от нормы информировать их не принято. Зато при повышении того или иного показателя, у ребенка в кратчайшие сроки возьмут кровь на повторный анализ.
В дальнейшем, если диагноз будет подтвержден, то наблюдение и лечение будет осуществлять специалист в профильном учреждении.
Неонатальный скрининг: прошлое и настоящее
Неонатальный скрининг сейчас — это простой, доступный и перспективный метод обследования новорожденных для выявления тяжелых наследственных заболеваний.
Его история началась в 1960-х годах в США с проведения биохимического теста на выявление фенилкетонурии.
Затем технология распространилась во многих странах мира и стала основой для создания перечня генетических болезней, которые можно диагностировать у детей в первые дни после рождения.
Существуют определенные критерии, по которым заболевание включается в перечень для скрининга в стране:
Пожалуй, последний пункт особенно важен. Ведь основная цель неонатального скрининга — не столько выявить заболевание, сколько начать вовремя лечение, чтобы обеспечить полноценную жизнь ребенку.
На сегодняшний день неонатальный скрининг проводится более чем в 50 странах мира, а перечень насчитывает более 50-60 заболеваний.
При домашних родах или ранней выписке из родильного дома обследование тоже проводят. И пройти его нужно обязательно, это в интересах и родителей, и малыша.
Неонатальный скрининг: 5 основных заболеваний
В большинстве регионов России проводится неонатальный скрининг на 5 основных генетических заболеваний.
- Фенилкетонурия — наследственное нарушение метаболизма аминокислот, в данном случае фенилаланина.
Характеризуется поражением центральной нервной системы. Единственный способ избежать тяжелых последствий — соблюдение диеты с низким содержанием фенилаланина уже с первой недели жизни ребенка.
- Адреногенитальный синдром, или врожденная дисфункция коры надпочечников, - наследственная ферментопатия. Развитие болезни обусловлено дефицитом стероидных гормонов (кортизола и альдостерона) и увеличением синтеза полового гормона — тестостерона.
Проявления заболевания разнообразны: от тяжелых нарушений водно-солевого обмена и полиорганной недостаточности до неправильного развития половых органов, маскулинизации и в дальнейшем бесплодия у девочек. В качестве лечения используется заместительная гормонотерапия.
- Врожденный гипотиреоз — неспособность щитовидной железы нормально развиваться или производить необходимое количество гормонов.
При отсутствии должного лечения происходит задержка психомоторного и речевого развития, наступает отставание в физическом и половом развитии. Показана пожизненная заместительная терапия препаратами тиреоидных гормонов.
- Галактоземия — нарушение углеводного обмена, при котором в организме накапливается избыток галактозы и ее метаболитов. Эти вещества обладают токсическим действием и повреждают различные органы и ткани.
Основная роль в лечении отводится диете с пожизненным исключением продуктов, содержащих лактозу и галактозу.
- Муковисцидоз — наследственное заболевание, при котором поражаются железы внешней секреции, а выделяемый ими секрет при этом становится чрезмерно густым и вязким.
Наиболее ярко болезнь проявляет себя со стороны органов дыхания — респираторная форма. Она дебютирует в раннем возрасте, проявляется частыми ОРВИ, бронхитами и пневмониями, сопровождается постоянным приступообразным кашлем с густой мокротой. В дальнейшем появляются и нарастают явления дыхательной и сердечной недостаточности.
Скрининг новорожденных входит в программу ОМС и проводится бесплатно.
Расширенный неонатальный скрининг
Современная технология диагностики — тандемная масс-спектрометрия — позволила на порядок расширить перечень наследственных заболеваний, которые можно включить в программу скрининга новорожденных: болезни обмена веществ, гемоглобинопатии, спинальная мышечная атрофия, тяжелый комбинированный иммунодефицит и другие.
В Москве с 2018 года дополнительно включены 6 заболеваний, отвечающих критериям Всемирной организации здравоохранения:
- глутаровая ацидурия тип 1;
- тирозинемия тип 1;
- лейциноз;
- метилмалоновая/пропионовая ацидурия (ацидемия);
- недостаточность биотинидазы;
- недостаточность ацил-КоА-дегидрогеназы среднецепочечных жирных кислот.
Все эти заболевания со столь сложными названиями относят к врожденным болезням обмена веществ (ВБО) или врожденным аномалиям метаболизма.
Причина их появления — дефекты единичных генов, которые кодируют ферменты, способствующие превращению одних веществ в другие. В результате таких нарушений метаболизма происходит накопление токсических веществ, воздействующих на различные органы и ткани, и отмечается дефицит важных веществ.
25% всех врожденных болезней обмена веществ дебютирует уже в период новорожденности. При этом они протекают тяжело, часто скрываются под маской других заболеваний и могут привести к смерти ребенка без должного лечения.
Неонатальный скрининг позволяет предупредить дебют опасной болезни и вовремя начать терапию.
Все ВБО контролируются путем соблюдения соответствующей диеты, ограничивающей поступление в организм веществ, обмен которых нарушен. Или, наоборот, путем введения в организм недостающих метаболитов.
До внедрения технологии тандемной масс-спектрометрии в практику неонатального скрининга в Москве было проведено исследование в Свердловской области в 2012-2014 годах. Тестирование прошли более 150 тысяч новорожденных детей на 16 врожденных болезней обмена веществ. Всего было выявлено 9 больных детей, что составляет 1 случай на 1130 детей.
Результат близок к средней по стране распространенности этих заболеваний. Исследование доказало эффективность нового метода диагностики и возможность его внедрения в другие регионы страны.
Тяжелая комбинированная иммунная недостаточность — место в неонатальном скрининге
Первичные врожденные иммунодефициты характеризуются неспособностью организма противостоять микробам и вирусам. Это неизбежно приводит к частым инфекционным заболеваниям, протекающим тяжело, длительно, с осложнениями и необходимостью раз за разом принимать антибиотики.
Наиболее опасной формой первичного иммунодефицита является тяжелая комбинированная иммунная недостаточность (ТКИН), при которой у детей резко снижено число лимфоцитов.
Заподозрить врожденный иммунодефицит в первые месяцы жизни ребенка сложно, ведь какое-то время болезнь может ничем себя не проявлять или имитировать симптомы других более распространенных патологий. Однако именно время установления диагноза во многом определяет успех лечения.
Наиболее эффективный метод лечения детей с тяжелой комбинированной иммунной недостаточностью — это трансплантация гемопоэтических стволовых клеток. В случае ее проведения в возрасте до 3,5 месяцев и при отсутствии инфекционных процессов отмечается хороший результат.
Неонатальный скрининг на ТКИН уже проводится в США, Израиле, некоторых странах Европы, Ближнего Востока и Азии.
Внесение ТКИН в программу неонатального скрининга в нашей стране позволит на ранних сроках диагностировать тяжелый врожденный иммунодефицит, вовремя проводить соответствующее лечение и предупреждать инфекционные осложнения.
Читайте также: