
Температура это в химии кратко
Обновлено: 08.07.2024
Температу́ра (от лат. temperatura — надлежащее смешение, нормальное состояние) — физическая величина, примерно характеризующая приходящуюся на одну степень свободы среднюю кинетическую энергию частиц макроскопической системы, находящейся в состоянии термодинамического равновесия.
В системе СИ температура измеряется в кельвинах. Но на практике часто применяют градусы Цельсия из-за привязки к важным характеристикам воды — температуре таяния льда (0° C) и температуре кипения (100° C). Это удобно, так как большинство климатических процессов, процессов в живой природе и т. д. связаны с этим диапазоном.
Существуют также шкалы Фаренгейта и некоторые другие.
Температура с молекулярно-кинетической точки зрения — физическая величина, характеризующая интенсивность хаотического, теплового движения всей совокупности частиц системы и пропорциональная средней кинетической энергии поступательного движения одной частицы.
Связь между кинетической энергией, массой и скоростью выражается следующей формулой:
Ek = 1/2m • v 2
Таким образом частицы одинаковой массы и имеющие одинаковую скорость имеют и одинаковую температуру.
Средняя кинетическая энергия частицы связана с термодинамической температурой постоянной Больцмана:
Eср = i/2kBT
где:
i — число степеней свободы
kB = 1.380 6505(24) × 10−23 Дж/K — постоянная Больцмана
T — температура;
Температура — величина, обратная изменению энтропии (степени беспорядка) системы при добавлении в систему единичного количества теплоты: 1/T = ΔS/ΔQ.
В равновесном состоянии температура имеет одинаковое значение для всех макроскопических частей системы. Если в системе два тела имеют одинаковую температуру, то между ними не происходит передачи кинетической энергии частиц (тепла) . Если же существует разница температур, то тепло переходит от тела с более высокой температурой к телу с более низкой, потому что суммарная энтропия при этом возрастает.
Свойства температуры изучает раздел физики — термодинамика. Температура также играет важную роль во многих областях науки, включая другие разделы физики, а также химию и биологию.
Если "на пальцах", то мера средней энергии частиц вещества. Если речь идёт о газе или жидкости - кинетической энергии, если о твёрдом веществе, тогда энергия колебаний частиц в решетке.
Тут важно, что это мера именно средней энергии, т. е. если частиц слишком мало, то понятие температуры теряет смысл. Например, в космосе: там всякие частицы носятся, но их слишком мало для того, чтобы усреднение энергий имело смысл.
Бобр в принципе верно написал, только колебания частиц в решетке -- это тоже кинетическая энергия. , так что самое краткое определение :
температура -- мера средней кинетической энергии структурных частиц вещества.
(от лат. temperatura- надлежащее смешение, нормальное состояние), термодинамич. параметр, характеризующий состояние термич. равновесия макроскопич. системы. Наряду с давлением, хим. потенциалом и др. параметрами состояния, Т. относится к интенсивным величинам, т. к. не зависит от массы системы. Согласно принципу термич. равновесия, две фазы А и В, адиабатически изолированные от окружающей среды (внутр. энергии фаз соотв. Е А + Е В =>const), могут находиться в состоянии равновесия, к-рое характеризуется определенными значениями и (р А, давления;, -молярные объемы фаз). Экспериментально установлено, что если фаза А находится в равновесии с фазой В, а В-с С, то А и С также находятся в равновесии. Из принципа термич. равновесия следует, что каждая фаза обладает эмпирической Т. q-измеримым св-вом такого рода, что из q А (р A;) = qB (р B;) и qB (р B;) = qC (р C;) следует q А(p А;) = q с (р с;).
Если две фазы с разл.q приведены в тепловой контакт друг с другом через пов-сть раздела и q А > q В, возникает поток теплоты от А к В, т. е. от более нагретой фазы к менее нагретой. При q А = q В тепловой поток отсутствует. Принцип термич. равновесия впервые сформулирован Дж. Блэком в кон. 18 в. В термодинамику он введен, однако, позднее первого и второго начал термодинамики, поэтому его часто называют нулевым началом термодинамики.
Существует множество ф-ций q(p,), удовлетворяющих нулевому началу. Конкретный вид q(p,) определяется используемым измерит. прибором-термометром и способом построения термометрич. шкалы.
Понятие абсолютной Т. введено У. Томсоном (лордом Кельвином) в 1848 на основании теоремы Карно, согласно к-рой все обратимые тепловые машины, где рабочее тело совершает круговой процесс между нагревателем с эмпирической Т. q1 и холодильником с эмпирической Т. q2, имеют одинаковый кпд h, независимо от природы рабочего тела:
где Q1- тепло, отбираемое рабочим телом от нагревателя, Q2 -тепло, передаваемое холодильнику. Значения q1 и q2 можно измерить с помощью произвольной шкалы Т., величины и h при смене шкалы остаются постоянными. Абсолютная Т. вводится соотношением:
где T1 и Т 2 -абсолютные Т. нагревателя и холодильника соотв., причем T1 есть ф-ция только q1, а Г 2 -только q2. Для построения шкалы абсолютной Т. достаточно приписать определенное значение Т, одному известному термич. состоянию. В настоящее время по международному соглашению принято, что абсолютная (термодинамич.) Т. плавления воды при нормальном давлении равна 273,15 К (точно). Абсолютный нуль Т. (или нулевая абсолютная Т.) имеют ясный физ. смысл как Т. холодильника в цикле Карно, при к-рой кпд тепловой машины h = 1. Тело, находящееся при нулевой Т., не способно передавать теплоту к.-л. другому телу. Единица измерения абсолютной Т. в системе СИ-градус Кельвина (Кельвин, К). Конкретные измерения абсолютной Т. осуществляются с помощью набора спец. термометров (подробнее см. Термометры, Термометрия).
Согласно строгой формулировке второго начала термодинамики (аксиоматика Каратеодори), абсолютная Т. вводится как интегрирующий делитель для бесконечно малого кол-ва теплоты dQ, полученного системой, обладающей внутренней энергией Е, в обратимом процессе. Величина dQ/T является полным дифференциалом ф-ции состояния S, наз. энтропией. Абсолютная Т. выражается соотношением:
X1, X2, Х3, . -экстенсивные термодинамич. переменные (объем V, электростатич. индукция D, магн. индукция Ви т. п.). Абсолютная Т. и эмпирическая Т. q связаны аналит. зависимостью для систем, у к-рых Eявляется ф-цией только Ти V:
Аналит. связь p,и Т для фазы наз. уравнением состояния. В статистической термодинамике аналогом ур-ния (1) служит соотношение:
где WЧ термодинамич. вероятность, k- постоянная Больц-мана. Термодинамич. вероятность W(E )равна числу возможных состояний системы, при к-рых последняя обладает внутр. энергией Е. Термодинамич. вероятность связана с энтропией соотношением Больцмана S= kln W. Для обычной макроскопич. системы величина W- быстро возрастающая ф-ция Еи, следовательно, абсолютная Т. положительна.
Термич. равновесие двух систем А и В (E А + Е B =const), определяемое равенством т-р T А = Т B, соответствует наиб. вероятному распределению энергии между А и В. Если В представляет собой обширный тепловой резервуар (Е В >> E А), то абсолютная Т. определяет плотность вероятности Р(Е) для системы А находиться в состоянии с заданной энергией EA,r при термич. равновесии с системой В:
где, суммирование ведется по всем значениям энергии EA.r (r =1,2. , п) подсистемы А (канонич. распределение Гиббса). Частными случаями канонич. распределения являются распределения молекул идеального газа по энергиям и скоростям (распределения Больцмана и Максвелла).
Важные физ. постоянные в-ва-его Т. кипения, плавления, фазовых переходов, полиморфных превращений, а также критическая Т. (см. Критическое состояние), тройные точки.
Практически все физ.-хим. величины зависят от Т. Важными примерами являются температурные зависимости:
1) константы скорости хим. р-ции к:
где A -энергия активации, A-предэкспоненциальный множитель (см. Аррениуса уравнение, Константа скорости, Энергия активации).
2) Константы равновесия хим. р-ции К р:
где R-газовая постоянная, -стандартная энтальпия р-ции.
3) Теплового эффекта хим. р-ции при постоянном давлении (DH) и постоянном объеме (DU):
где H и U-энтальпия и внутр. энергия системы, С р и С V-> теплоемкости при постоянном давлении и постоянном объеме соотв. (см. Кирхгофа уравнение).
4) Теплоты равновесного фазового перехода L:
где -изменение молярного объема при переходе в-ва из фазы 1 в фазу 2 (см. Клапейрона -Клаузиуса уравнение).
5) Стандартной электродвижущей силы E 0 гальванич. цепи:
где К р (Т)-константа равновесия электродного процесса, F-Фарадея постоянная, Z -число переносимых электронов (см. Электрохимические цепи).
6) Объемной плотности rv излучения абсолютно черного тела с частотой v (ф-ла Планка):
где с-скорость света, h-постоянная Планка.
Полной объемной плотности излучения по всем частотам (закон Стефана - Больцмана):
7) Степени ионизации a газа, состоящего из атомов А:
где i -энергия ионизации атома, m-масса электрона; i , g> А -статистич. веса ионов и атомов (ур-ние Саха).
Понятие Т., сформулированное для равновесного состояния системы в целом, используется и для характеристики локального термодинамич. равновесия, если система в целом неравновесна и ее Т. рассматривается как непрерывная ф-ция координат и времени. При локальном термодинамич. равновесии малые элементы объема приближенно рассматриваются как равновесные, обладающие каждый своей Т., и учитывается обмен энергией (энтропией) между ними. Локальное термодинамич. равновесие-одно из осн. понятий термодинамики необратимых процессов. В ряде физ. задач неравновесная система м. б. разбита на подсистемы, в к-рых время установления термич. равновесия много меньше времени достижения равновесия системой в целом. Подобная ситуация м. б. охарактеризована тем, что каждой из подсистем соотносится своя Т., отличная от Т. других подсистем. Напр., в полупроводниках Т. электронов проводимости в сильном электрич. поле много выше Т. решетки; в плазме отдельно рассматривают Т. электронов и Т. ионов.
В нач. 50-х гг. 20 в. сформулировано понятие отрицательных абсолютных Т. Такие Т. могут возникать в системах, если с ростом энергии Етермодинамич. вероятность W(или энтропия S)не возрастает, а убывает, в результате чего производная становится меньше нуля (см. ф-лы 1 и 2). Подобная ситуация реализуется для таких систем, в к-рых энергия Еограничена снизу и сверху. Так, двухуровневая система, состоящая из Nядерных спинов во внеш. магн. поле (напр., ионы Li + в кристалле LiF), имеет миним. энергию NE1, максимальную NE2, где E1 и Е 2 -энергии спина ядра на нижнем и верхнем уровнях. Начиная с энергии, равной N(E1 + E2)/2, термодинамич. вероятность Wубывает с ростом энергии, что позволяет говорить об отрицательной Т. подсистемы (ионы Li + ), но не для системы в целом. Рассматриваемая подсистема должна быть термически слабо связана с системой в целом, для к-рой отсутствуют ограничения по энергии.
При физ.-хим. исследованиях условно выделяют область низких Т. (см. Криохимия) и область высоких Т. (обычно 500-3000 К), к-рую рассматривают как химию высоких Т., или просто высокотемпературную химию. Т. в интервале 500-3000 К получают методами радиационного и лазерного нагрева, электронной и ионной бомбардировки. Объекты высокотемпературной химии, как правило,-неорг. соединения. Характерными чертами высокотемпературных хим. процессов являются: 1) сравнительно малая роль констант скорости, энергий активации и т. п. кинстич. факторов, поскольку скорость р-ций высока и в системе быстро устанавливается равновесие; 2) увеличение роли газовой (паровой) фазы из-за интенсивных процессов испарения; 3) необходимость учета влияния заряженных частиц-ионов и электронов, возникающих в результате термодиссоциации (см. Ионы в газах, Ионно-молекулярные реакции).Высокотемпературными процессами являются мн. металлургич. произ-ва, процессы напыления пленок, монокристаллов выращивания из газовой фазы и др.
Процессы в области Т. 3000-5000 К изучаются плазмо-химией.
Лит.: Кричевский И. Р., Понятия и основы термодинамики, 3 изд., М., 1962; Рей Ф., Статистическая термодинамика, пер. о англ., М., 1986.
М. В. Коробов.
высокая температура, жар, криотемпература, ликвидус, лямбда-точка, палеотемпература, повышенная температура, субфебрилитет
Существует два определения температуры. Одно - с молекулярно-кинетической точки зрения, другое - с термодинамической.
В системе СИ температура измеряется в воды — температуре таяния льда (0° C) и температуре кипения (100° C). Это удобно, так как большинство климатических процессов, процессов в живой природе и т. д. связаны с этим диапазоном.
Существуют также шкалы другие .
Содержание
Молекулярно-кинетическое определение
Температура с молекулярно-кинетической точки зрения — физическая величина, характеризующая интенсивность хаотического, теплового движения всей совокупности частиц системы и пропорциональная средней кинетической энергии поступательного движения одной частицы.
Связь между кинетической энергией, массой и скоростью выражается следующей формулой:
Ek = 1 /2m • v 2
Таким образом частицы одинаковой массы и имеющие одинаковую скорость имеют и одинаковую температуру.
Средняя кинетическая энергия частицы связана с термодинамической температурой Термодинамическое определение
Температура — величина, обратная изменению энтропии (степени беспорядка) системы при добавлении в систему единичного количества теплоты: 1/T = ΔS/ΔQ.
История термодинамического подхода
Измерение температуры
Для измерения температуры выбирается некоторый термодинамический параметр термометрического вещества. Изменение этого параметра однозначно связывается с изменением температуры. На практике для измерения температуры используют
-
Единицы и шкала измерения температуры
Из того, что температура — это кинетическая энергия молекул, ясно, что наиболее естественно измерять её в энергетических единицах (т.е. в системе СИ в Шкала температур Кельвина
Абсолютный ноль определён как 0 K, что приблизительно равно −273.15 °C.
Шкала температур Кельвина — Шкала Цельсия
В быту используется нормальном атмосферном давлении . Поскольку температура замерзания и кипения воды недостаточно хорошо определена, в настоящее время шкалу Цельсия определяют через шкалу Кельвина: градус Цельсия равен кельвину, абсолютный ноль принимается за −273,15° C. Шкала Цельсия практически очень удобна, поскольку вода очень распространена на нашей планете и на ней основана наша жизнь. Ноль Цельсия — особая точка для Андерсом Цельсием в 1742 г.
Шкала Фаренгейта
В Англии и, в особенности, в США используется шкала Фаренгейта. Ноль градусов Цельсия — это 32 градуса Фаренгейта, а градус Фаренгейта равен 5/9 градуса Цельсия.
В настоящее время принято следующее определение шкалы Фаренгейта: это температурная шкала, 1 градус которой (1 °F) равен 1/180 разности температур кипения воды и таяния льда при атмосферном давлении, а точка таяния льда имеет температуру +32 °F. Температура по шкале Фаренгейта связана с температурой по шкале Цельсия (t °С) соотношением t °С = 5/9 (t °F - 32), 1 °F = 9/5 °С + 32. Предложена Г. Фаренгейтом в 1724.
Энергия теплового движения при абсолютном нуле
Температура с термодинамической точки зрения
Существует множество различных шкал температур. Когда-то температура определялась очень произвольно. Мерой температуры служили метки, нанесённые на равных расстояниях на стенах трубочки, в которой при нагревании расширялась вода. Потом решили измерить температуру , которая не зависит от свойств вещества. Из термодинамики следует, что если какая-то тепловая машина, поглощая количество теплоты
при

выделяет тепло
при температуре в один при
, выделяет то же самое тепло
при температуре в один градус, то машина, поглощающая
при
должна при температуре
выделять тепло
. Конечно, между теплом
и температурой
существует зависимость и тепло
должно быть пропорционально
. Таким образом, каждому количеству тепла
, выделенного при температуре в один градус, соответствует количество тепла, поглощённого машиной при температуре
, равное
, умноженному на некоторую возрастающую функцию
температуры:
Поскольку найденная функция возрастает с температурой, то можно считать, что она сама по себе измеряет температуру, начиная со стандартной температуры в один градус. Это означает, что можно найти температуру тела, определив количество тепла, которое поглощается тепловой машиной, работающей в интервале между температурой тела и температурой в один градус. Полученная таким образом температура называется абсолютной термодинамической температурой и не зависит от свойств вещества. Таким образом, для обратимой тепловой машины выполняется равенство:
— энтропия:
Для системы, в которой энтропия
может быть функцией
её энергии
, термодинамическая температура определяется как:
Температура и излучение
При повышении температуры растёт энергия, излучаемая нагретым телом. Энергия излучения абсолютно чёрного тела описывается законом Стефана — Больцмана
Шкала Реомюра
Предложена в Переходы из разных шкал
| в\из | Кельвин | Цельсий | Фаренгейт |
|---|---|---|---|
| Сравнение температурных шкал |
¹ Нормальная средняя температура человеческого тела — 36.6 ° C ±0.7 ° C, или 98.2 °F ±1.3 °F. Приводимое обычно значение 98.6 °F - это точное преобразование в шкалу Фаренгейта принятого в Германии в XIX веке значения 37 ° C. Однако это значение не входит в диапазон нормальной средней температуры тела человека, поскольку температура разных частей тела разная [1] .
Накопительный водонагреватель
Накопительный водонагреватель, или бойлер (от англ. boiler) представляет собой сравнительно большую ёмкость с размещенным в ней или, реже, под ней, источником тепла. Нагрев может производиться при помощи парового или водяного теплообменника — в нём циркулирует горячая вода в замкнутом контуре, нагреваемая, например, с помощью отопительного котла. Такие бойлеры называют бойлерами (водонагревателями) косвенного нагрева. [2]
Некоторые значения в этой таблице были округлены.
Характеристика фазовых переходов
Для описания точек фазовых переходов различных веществ используют следующие значения температуры:
Измерение температуры
Для измерения температуры выбирается некоторый термодинамический параметр термометрического вещества. Изменение этого параметра однозначно связывается с изменением температуры. На практике для измерения температуры используют
ТЕМПЕРАТУРА (от лат. temperatura- надлежащее смешение, нормальное состояние), термодинамич. параметр, характеризующий состояние термич. равновесия макроскопич. системы. Наряду с давлением, хим. потенциалом и др. параметрами состояния, температура относится к интенсивным величинам, т.к. не зависит от массы системы. Согласно принципу термич. равновесия, две фазы А и В, адиабатически изолированные от окружающей среды (внутр. энергии фаз соотв. Е А + Е В = const), могут находиться в состоянии равновесия, к-рое характеризуется определенными значениямии(р А , р B -давления;, -молярные объемы фаз). Экспериментально установлено, что если фаза А находится в равновесии с фазой В, а В-с С, то А и С также находятся в равновесии. Из принципа термич. равновесия следует, что каждая фаза обладает эмпирической температурой q -измеримым св-вом такого рода, что из q А (р A ;) = q B (р B ;) и q B (р B ;) = q C (р C ;) следует q А (p А ;) = q с (р с ;).
Если две фазы с разл. q приведены в тепловой контакт друг с другом через пов-сть раздела и q А > q В , возникает поток теплоты от А к В, т. е. от более нагретой фазы к менее нагретой. При q А = q В тепловой поток отсутствует. Принцип термич. равновесия впервые сформулирован Дж. Блэком в кон. 18 в. В термодинамику он введен, однако, позднее первого и второго начал термодинамики, поэтому его часто называют нулевым началом термодинамики.
Существует множество ф-ций q (p,), удовлетворяющих нулевому началу. Конкретный вид q (p,) определяется используемым измерит. прибором-термометром и способом построения термометрич. шкалы.
Понятие абсолютной температуры введено У. Томсоном (лордом Кельвином) в 1848 на основании теоремы Карно, согласно к-рой все обратимые тепловые машины, где рабочее тело совершает круговой процесс между нагревателем с эмпирической температурой q 1 и холодильником с эмпирической температурой q 2 , имеют одинаковый кпд h , независимо от природы рабочего тела:
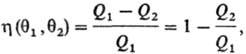
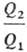
где Q 1 - тепло, отбираемое рабочим телом от нагревателя, Q 2 -тепло, передаваемое холодильнику. Значения q 1 и q 2 можно измерить с помощью произвольной шкалы температуры, величиныи h при смене шкалы остаются постоянными. Абсолютная температура вводится соотношением:
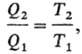
где T 1 и Т 2 -абсолютные температуры нагревателя и холодильника соотв., причем T 1 есть ф-ция только q 1 , а T 2 -только q 2 . Для построения шкалы абсолютной температуры достаточно приписать определенное значение Т, одному известному термич. состоянию. В настоящее время по международному соглашению принято, что абсолютная (термодинамич.) Температура плавления воды при нормальном давлении равна 273,15 К (точно). Абсолютный нуль температуры (или нулевая абсолютная температура) имеют ясный физ. смысл как температура холодильника в цикле Карно, при к-рой кпд тепловой машины h = 1. Тело, находящееся при нулевой температуре, не способно передавать теплоту к.-л. другому телу. Единица измерения абсолютной температуры в системе СИ-градус Кельвина (Кельвин, К). Конкретные измерения абсолютной температуры осуществляются с помощью набора спец. термометров (подробнее см. Термометры, Термометрия).
Согласно строгой формулировке второго начала термодинамики (аксиоматика Каратеодори), абсолютная температура вводится как интегрирующий делитель для бесконечно малого кол-ва теплоты d Q, полученного системой, обладающей внутренней энергией Е, в обратимом процессе. Величина d Q/T является полным дифференциалом ф-ции состояния S, наз. энтропией. Абсолютная температура выражается соотношением:
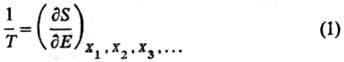
X 1 , X 2 , Х 3 , . -экстенсивные термодинамич. переменные (объем V, электростатич. индукция D, магн. индукция В и т.п.). Абсолютная температура и эмпирическая температура q связаны аналит. зависимостью для систем, у к-рых E является ф-цией только Ти V:
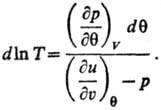
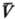
Аналит. связь p,и Т для фазы наз. уравнением состояния. В статистической термодинамике аналогом ур-ния (1) служит соотношение:
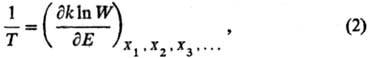
где W— термодинамич. вероятность, k-постоянная Больц-мана. Термодинамич. вероятность W(E)равна числу возможных состояний системы, при к-рых последняя обладает внутр. энергией Е. Термодинамич. вероятность связана с энтропией соотношением Больцмана S = kln W. Для обычной макроскопич. системы величина W- быстро возрастающая ф-ция Е и, следовательно, абсолютная температура положительна.
Термич. равновесие двух систем А и В (E А + Е B = const), определяемое равенством т-р T А = Т B , соответствует наиб. вероятному распределению энергии между А и В. Если В представляет собой обширный тепловой резервуар (Е В >> E А ), то абсолютная температура определяет плотность вероятности Р(Е) для системы А находиться в состоянии с заданной энергией E A,r при термич. равновесии с системой В:
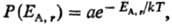
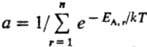
где, суммирование ведется по всем значе ниям энергии E A.r (r = 1,2. , п) подсистемы А (канонич. распределение Гиббса). Частными случаями канонич. распределения являются распределения молекул идеального газа по энергиям и скоростям (распределения Больцмана и Максвелла).
Практически все физ.-хим. величины зависят от температуры. Важными примерами являются температурные зависимости:
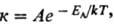
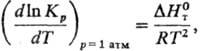
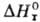
где R-газовая постоянная, -стандартная энтальпия р-ции.
3) Теплового эффекта хим. р-ции при постоянном давлении ( D H) и постоянном объеме ( D U):
( 9 D Н/ 9 Т) р = D С р , ( 9 D U/ 9 Т) V = D С V ,
где H и U-энтальпия и внутр. энергия системы, С р и С V -теплоемкости при постоянном давлении и постоянном объеме соотв. (см. Кирхгофа уравнение).
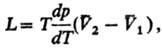
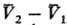
где -изменение молярного объема при переходе в-ва из фазы 1 в фазу 2 (см. Клапейрона -Клаузиуса уравнение).
5) Стандартной электродвижущей силы E 0 гальванич. цепи:
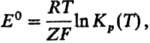
6) Объемной плотности r v излучения абсолютно черного тела с частотой v (ф-ла Планка):
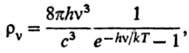
где с-скорость света, h-постоянная Планка.
Полной объемной плотности излучения по всем частотам (закон Стефана - Больцмана):
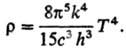
7) Степени ионизации a газа, состоящего из атомов А:
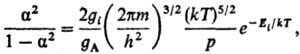
где E i -энергия ионизации атома, m-масса электрона; g i , g А -статистич. веса ионов и атомов (ур-ние Саха).
Понятие температуры, сформулированное для равновесного состояния системы в целом, используется и для характеристики локального термодинамич. равновесия, если система в целом неравновесна и ее температура рассматривается как непрерывная ф-ция координат и времени. При локальном термодинамич. равновесии малые элементы объема приближенно рассматриваются как равновесные, обладающие каждый своей температурой, и учитывается обмен энергией (энтропией) между ними. Локальное термодинамич. равновесие-одно из осн. понятий термодинамики необратимых процессов. В ряде физ. задач неравновесная система м. б. разбита на подсистемы, в к-рых время установления термич. равновесия много меньше времени достижения равновесия системой в целом. Подобная ситуация м. б. охарактеризована тем, что каждой из подсистем соотносится своя температура, отличная от температур других подсистем. Напр., в полупроводниках температура электронов проводимости в сильном электрич. поле много выше температуры решетки; в плазме отдельно рассматривают температуру электронов и температуру ионов.
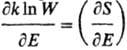
В нач. 50-х гг. 20 в. сформулировано понятие отрицательных абсолютных температур. Такие температуры могут возникать в системах, если с ростом энергии Е термодинамич. вероятность W (или энтропия S)не возрастает, а убывает, в результате чего производная становится меньше нуля (см. ф-лы 1 и 2). Подобная ситуация реализуется для таких систем, в к-рых энергия Е ограничена снизу и сверху. Так, двухуровневая система, состоящая из N ядерных спинов во внеш. магн. поле (напр., ионы Li + в кристалле LiF), имеет миним. энергию N E 1 , максимальную NE 2 , где E 1 и Е 2 -энергии спина ядра на нижнем и верхнем уровнях. Начиная с энергии, равной N (E 1 + E 2 )/2, термодинамич. вероятность W убывает с ростом энергии, что позволяет говорить об отрицательной температуре подсистемы (ионы Li + ), но не для системы в целом. Рассматриваемая подсистема должна быть термически слабо связана с системой в целом, для к-рой отсутствуют ограничения по энергии.
При физ.-хим. исследованиях условно выделяют область низких температур (см. Криохимия) и область высоких температур (обычно 500-3000 К), к-рую рассматривают как химию высоких температур, или просто высокотемпературную химию. Температуры в интервале 500-3000 К получают методами радиационного и лазерного нагрева, электронной и ионной бомбардировки. Объекты высокотемпературной химии, как правило,-неорг. соединения. Характерными чертами высокотемпературных хим. процессов являются: 1) сравнительно малая роль констант скорости, энергий активации и т. п. кинстич. факторов, поскольку скорость р-ций высока и в системе быстро устанавливается равновесие; 2) увеличение роли газовой (паровой) фазы из-за интенсивных процессов испарения; 3) необходимость учета влияния заряженных частиц-ионов и электронов, возникающих в результате термодиссоциации (см. Ионы в газах, Ионно-молекулярные реакции). Высокотемпературными процессами являются мн. металлургич. произ-ва, процессы напыления пленок, монокристаллов выращивания из газовой фазы и др.
Процессы в области температур 3000-5000 К изучаются плазмохимией.
Лит.: Кричевский И. Р., Понятия и основы термодинамики, 3 изд., М., 1962; Рей Ф., Статистическая термодинамика, пер. о англ., М., 1986.
Читайте также: