
Синдром ретта лечение кратко
Обновлено: 05.07.2024
Распространенность
Что провоцирует / Причины Синдрома Ретта:
Анализ морфологических изменений при синдроме Ретта указывает на замедление развития мозга после рождения и остановку его роста к 4-летнему возрасту. Выявлено замедление роста тела и отдельных органов (сердца, печени, почек, селезенки).
Симптомы Синдрома Ретта:
В течении заболевания выделяют 4 стадии:
I стадия (возраст ребенка 6-12 мес.): слабость мышечного тонуса, замедление роста в длину кистей, стоп, окружности головы.
II стадия (возраст 12-24 мес.): атаксия туловища и походки, машущие и подергивающие движения рук, необычные перебирания пальцами.
III стадия: утрата ранее приобретенных навыков, способности к игре, коммуникациям (в том числе визуальным).
IV стадия: распад речи, возникновение эхолалий (в том числе ретардированных), неправильное употребление местоимений.
Первая стадия - стагнация. Включает замедление психомоторного развития ребенка, замедление роста головы, потерю интереса к играм, диффузную мышечную гипотонию.
После фазы регресса наступает третья стадия - псевдостационарная, охватывающая длительный период дошкольного и раннего школьного возраста. Состояние детей относительно стабильно. На первый план выступают глубокая умственная отсталость, судорожные припадки, экстрапирамидные расстройства по типу мышечной дистонии, атаксии, гиперкинезов. Приступов беспокойства не отмечается.
В конце первого десятилетия жизни начинается четвертая стадия - прогрессирования двигательных нарушений. Больные становятся обездвиженными, нарастают спастичность, мышечные атрофии, вторичные деформации - сколиоз, появляются вазомоторные расстройства преимущественно в нижних конечностях. Характерно отставание в росте без задержки полового созревания. Имеется тенденция к развитию кахексии. Судорожные приступы редкие. У больных с синдромом Ретта на фоне тотального распада всех сфер деятельности наиболее длительно сохраняются эмоциональное общение и привязанности, соответствующие уровню их психического развития.
Диагностика Синдрома Ретта:
Диагностические критерии синдрома Ретта по Е. Trevathan
- 1) дыхательные расстройства (периодическое апное во время бодрствования, перемежающееся гипервентиляцией, аэрофагия);
- 2) судорожные припадки;
- 3) спастичность, часто сочетающаяся с дистонией и атрофией мышц;
- 4) сколиоз;
- 5) задержка роста;
- 6) гипотрофичные маленькие ступни;
- 7) ЭЭГ аномалии (медленный фоновый ритм и периодическое замедление ритма до 3-5 Гц, описаны центральновисочные Spikes как при фрагильной Х-хромосоме и роландической эпилепсии).
Дифференциальная диагностика
Дифференциальный диагноз синдрома Ретта и раннего детского аутизма (РДА) (Международная конференция по синдрому Ретта, 1988)

Синдром Ретта – это генетическое заболевание, сопровождающееся тяжелыми психоневрологическими симптомами. Диагностика его затруднительна: оно практически никогда не обнаруживается внутриутробно, а после рождения проявляется не ранее, чем через 6 месяцев. Своему носителю оно грозит глубоким слабоумием, двигательными ограничениями, дезадаптацией в социуме.
Истоки заболевания
В масштабном формате о расстройстве заговорили в 1983 году благодаря шведскому ученому Бенгту Хагбергу. В это время он со своей группой изучал 35 подобных между собой случаев в 3 разных странах: в Португалии, Франции и Швеции.
Однако Хагберт не является первооткрывателем синдрома. Впервые его обнаружил педиатр Андреас Ретт, имя которого носит заболевание. Он наблюдал за двумя девочками, имеющими одинаковые симптомы. Их он заметил в очереди на прием. Они сидели на коленях у матерей, а те держали их за руки. Девочки раскачивались как маятники, а затем внезапно обе начали совершать стереотипные движения руками. Дети застыли в одном положении, отстраненные от окружающего мира. Взгляд был направлен в одну точку. Поражала их синхронность в движениях и поведении.
В своих письменных архивах врач отыскал подобные истории болезни, а затем отправился в Европу, чтобы разыскать и там таких же пациентов. В 1966 он сделал первые публикации своих исследований, которые, однако, не вызвали особого интереса.
Зафиксированную им болезнь Ретт назвал синдромом атрофии мозга. Сначала ее считали проявлением аутизма или шизофрении, и только лишь в 1983 году вывели в отдельную нозологическую единицу.

В настоящее время синдром относят к категории довольно редких генетических заболеваний. Он встречается с частотой случаев 1 на 15000. Причиной его называют мутацию гена МЕСР2. Этот ген отвечает за синтез определенного белка, влияющего на развитие мозга. В норме этот белок, спустя некоторое время после рождения, должен подавляться другими генами, чтобы обеспечить нормальное развитие мозга.
Если же ген МЕСР2 мутирован, то белок инактивируется не полностью, что вызывает аномальное мозговое созревание, и провоцирует развитие синдрома Ретта.
Обычно мутирующий ген располагается в Х хромосоме, потому заболеванием страдают преимущественно девочки.
Почему мальчики не болеют
У мальчика Х-хромосома одна. Если она имеет мутационный ген, значит, выпадает из работы полностью, и ее нечем заменить. Такие малыши мужского пола, как правило, погибают еще внутриутробно, так и не родившись. Поэтому синдром Ретта у мальчиков встречается крайне редко.

Но, несмотря на такую особенность заболевания, очень редко, но все-таки мальчики с подобным синдромом выживают. Это может быть связано с тем, что не все гены в Х-хромосоме подвергаются мутации. Из-за этого заболевание развивается не столь остро.
Другая причина – наличие у мальчика синдрома Клайнфельтера. При этом наблюдается полисомия половых хромосом, то есть их набор составляет ХХУ. И, если одна Х-хромосома имеет патологический ген, то вторая может регулировать синтез белка и дарить мальчику возможность жизни. Получается такая же картина, как и у девочки.
Как развивается заболевание
Синдром Ретта у детей – довольно коварное заболевание. При рождении оно практически не проявляет себя. Первые его симптомы появляются в период от 6 мес. до полутора лет. Однако некоторые, еле заметные признаки, в первом полугодии все-таки имеются. Но они настолько ничтожны, что не привлекают внимания.
В 1 год и 7 мес. она перестала узнавать родителей и, казалось, не нуждалась в них. Весь день проводила за одним однообразным занятием: кидала мяч или катала коляску. Часами ходила по кругу, пока ее не останавливали или она запиналась. Такое стереотипное поведение носит название полевого, когда действие затягивает больного, и он не может ничего сделать.

В четыре года к симптомам присоединились эпилептоидные припадки. Однако по достижении школьного возраста девочка находилась на домашнем обучении, и делала некоторые успехи.
12–6 лет – это был период ремиссии, когда болезнь практически не беспокоила. Но с 16 лет появились новые, более глубокие проблемы, связанные с костными деформациями и болезнями внутренних органов. Одна нога девочки была короче другой почти на 10 см, что не могло не препятствовать ходьбе. В 20 лет она весила всего 24 кг с ростом 158 см.
Обычно СР протекает в 4 стадии.
Первая стадия, которая, как правило, стартует с 6 месяцев до полутора лет, проявляется нарастанием раздражительности и лабильностью настроения у ребенка. Эпизоды плача и психомоторного возбуждения сменяются все большей пассивностью. Малыш бесцельно передвигается по комнате, пропадает интерес к игрушкам. Но контакт с матерью сохраняется.
Вот как описывает женщина поведение своей дочери на заре заболевания: она кричала целый день без остановки, билась головой о стены, не могла уснуть. Что бы мы ни делали, она не успокаивалась. Это был настоящий ад. Но больше угнетало то, что ни один врач не мог поставить вразумительный диагноз.
Развивается диспропорция головы и конечностей по отношению к телу. Они становятся несоизмеримо маленькими. Замедляется рост, и снижается тонус мышц.
Вторая стадия, длящаяся несколько лет, отличается пестротой симптомов. Сразу обращает на себя внимание снижение интеллектуальных способностей, развивается умственное слабоумие. Происходит регресс практически всех полученных навыков. Речь полностью исчезает или переходит в степень эхолалии – механического повторения услышанного.
Приобретенные двигательные навыки, предметно-ролевое поведение теряются и замещаются двигательными стереотипами. Характерный симптом: многочисленно повторяющиеся движения, напоминающие мытье рук. Кроме этого, ребенок постоянно заламывает или потирает их, размахивает ими, хлопает в ладоши. Сжатие пальцев рук вполне нормально в 4 месяца, но в более позднем возрасте говорит об остановке развития. Малыш утрачивает хватательный рефлекс, не способен производить вращательные движения руками.
Постепенно двигательная активность сходит на нет. Нарушается походка, ребенок ходит, не сгибая коленей.

Третья стадия длится 10 лет и более, характеризуется она развитием стойкого, глубокого слабоумия, вплоть до идиотии. Наблюдается полная потеря способности говорить и понимать обращенную к ребенку речь. Появляется тремор всего тела, отягчающий движения. Усиливаются судорожные припадки.
Четвертая, конечная стадия – это период усугубления ранее проявляемых симптомов. Стойкая утрата умственных способностей, двигательных навыков, развитие мышечных дистрофий, приводящих к полному обездвиживанию.
Продолжительность жизни таких больных в среднем колеблется до 30 лет, хотя известны случаи, когда они доживали и до 50-летнего возраста.
Самые частые симптомы расстройства
Типичные симптомы для синдрома Ретта – мышечные и двигательные нарушения. Мышцы находятся в гипертонусе или же, наоборот, теряют его. В этом случае у ребенка развивается неправильное положение тела, прогрессирует частичные параличи и нарушение координации. Например, девочки скрещивают ноги во время ходьбы.
Синкинезии – патологические сокращения мышц, возникают вслед за произвольным движением: простая улыбка способна вызвать резкий взмах ногой. Такое явление постепенно приводит к повреждению суставов, сухожилий и связок, провоцирует ортопедические нарушения. Последние проявляются во всевозможных деформациях и также очень часто сопровождают таких детей. Среди них выделяют вывих тазобедренного сустава, провоцируемый малой подвижностью.
Сколиоз – боковое искривление позвоночника, который провоцирует массу проблем у таких пациентов: деформации суставов и костей, боли во время ходьбы, в стоячем или сидячем положении, утрата способности передвигаться. Сколиоз грудного отдела вызывает легочную недостаточность. Появляются также проблемы с пищеварением.
У детей с синдромом Ретта наблюдается повышенное слюнотечение. Но это происходит не из-за избытка количества слюны, а потери способности сглатывать ее.
Нарушение питания может развиваться из-за частых приступов тошноты. Она появляется на любые аспекты питания: на определенный продукт, его температуру, на способ приготовления. Так, ребенок способен отрицательно реагировать на пищу, поданную кусочками, или на комочки в блюде.

Постоянная тошнота провоцирует отказ от питания, а значит, потерю в весе.
Плохое сглатывание слюны, которая регулирует кислотность в желудке, и повышенное внутрибрюшное давление вызывают желудочно-пищеводный рефлюкс, то есть забрасывание содержимого желудка в пищевод. Это чревато такими последствиями, как воспаление стенки пищевода, респираторные инфекции.
Малоподвижный образ жизни, неврологические расстройства, неправильное питание провоцируют возникновение запоров у детей с синдромом Ретта. Они носят тяжелый характер, поскольку способны вызывать закупорку кишечника и сильные боли.
Повышенное слюнотечение, тошнота, рефлюкс снижают потребление ребенком пищи и даже развивают на нее негативную реакцию. В результате этого ребенок теряет в весе. Этот процесс стоит строго контролировать, поскольку он чреват истощением.
Другое тяжелое расстройство связано с работой дыхательной системы, развивающееся вплоть до приступов апноэ. Это явление настолько часто среди детей с синдромом, что нередко стает причиной их гибели.
Важными патогномоничными признаками синдрома считаются проявления аутизма. Именно из-за них заболевание изначально считали одной из форм этого расстройства, а в настоящее время относят к болезням аутистического спектра.
Аутистические признаки проявляются в отстранении от окружающего мира, в том числе и от родственников. Ребенок замыкается в себе, может не откликаться, когда его зовут. Предпочитает одиночество. Дети боятся чужих людей и непривычных ситуаций.
Лицо такого ребенка становится похожим на каменное. Взгляд блуждающий или устремлен в одну точку. Поведение часто непредсказуемо: случаются приступы неутомимого смеха или плача. Склонны к самоповреждениям: царапают кожу, кусают пальцы, вырывают волосы.
Нетипичная картина
Наряду с типичной формой заболевания, описанной выше, встречаются и атипичные формы. Они имеют свои особенности, от которых зависит тяжесть заболевания.
- Zapella – форма синдрома с неярко выраженными признаками. Речь частично сохранена, умеренно выражен сколиоз, умственная отсталость средней степени тяжести. Физически развиваются нормально.
- Hanefeld – в клинической картине преобладает раннее развитие судорожных приступов. Часто они случаются даже до появления умственной деградации.
- Rolando – на первый план выходят признаки задержки психомоторного развития. Ребенок теряет возможность передвигаться, нарастает стереотипия движений, его беспокоят дыхательные нарушения.

Синдром Ретта – сложное генетическое заболевание. Прежде всего, его сопровождает полная умственная деградация и психоневрологические нарушения, влекущие за собой многочисленные патологии других систем организма.
К сожалению, в мире еще не существует способа кардинального искоренения болезни, хотя ученые ведут постоянные разработки в этом направлении.
Лечение синдрома сводится к трем основным направлениям. Медикаментозная терапия назначается для купирования судорожных припадков и стимуляции работы головного мозга.
Диетотерапия включает в себя контроль массы тела, употребление в пищу высококалорийных, витаминизированных продуктов.
Однако наибольшее внимание уделяется реабилитационным мероприятиям, направленным на укрепление опорно-двигательного аппарата и поддержание умственного, психомоторного развития.
Важно сохранить комплексный, всесторонний подход к проблеме. Такие дети нуждаются в постоянной поддержке со стороны взрослых и веры в них. Сотрудничество с ними, как с полноценной ячейкой общества, способствует их лучшей адаптации в социуме и более благоприятному развитию.
Синдром Ретта: причины, диагностика, лечение
Этиология и встречаемость синдрома Ретта. Синдром Ретта (MIM № 312750) — панэтническое Х-сцепленное доминантное заболевание с распространением среди девочек 1 на 10 000-15 000.
Вызывается мутациями с утратой функции в гене МЕСР2. Описано несколько мальчиков с выраженными нарушениями развития и неврологическими аномалиями с мутациями, вызывающими частичную потерю функции МЕСР2, но обычно у мужчин типичного синдрома Ретта не бывает, кроме случаев кариотипа 47.XXY или соматического мозаицизма.
У нескольких пациентов с атипическим синдромом Ретта найдены мутации в одном, также Х-сцепленном, аллеле гена CDKL5. Белок CDKL5 — киназа треонина и серина, но о его функции мало известно.
Патогенез синдрома Ретта
Ген МЕСР2 кодирует ядерный белок, связанный с метилированием ДНК и переносящий гистоновую деацетилазу в область метилирования ДНК. Точная функция МеСР2 полностью не определена, но существует гипотеза, что он связан с транскрипционным молчанием и эпигенетической регуляцией генов в областях метилированной ДНК. Соответственно дисфункция или утрата МеСР2, наблюдаемая при синдроме Ретта, должна вызывать неправильную активизацию гена.
Мозг у пациентов с синдромом Ретта небольшого размера, с атрофией коры и мозжечка, но без потери нейронов; синдром Ретта, следовательно, не относится к типичным нейродегенеративным заболеваниям. В коре и гиппокампе нейроны пациентов с синдромом Ретта имеют меньшие размеры и более плотно упакованы, чем в норме, и имеют упрощенное ветвление дендритов.
Эти наблюдения указывают, что белок МеСР2 важен для возникновения и поддержки межнейронного взаимодействия, а не для пролиферации предшественников нейронов или их дифференцировки.
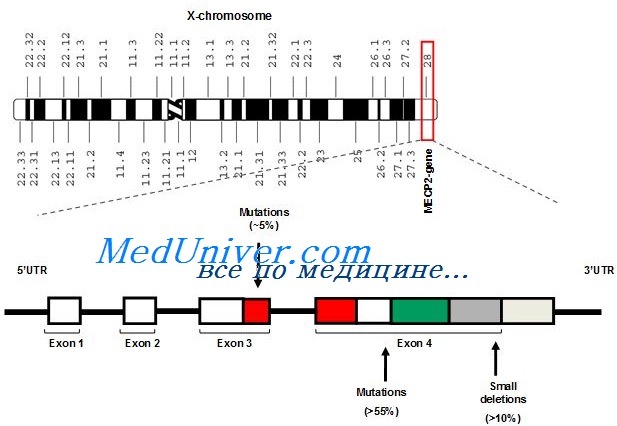
Фенотип и развитие синдрома Ретта
Впоследствии они быстро теряют речь и приобретенные двигательные навыки, особенно целенаправленного использования рук. В ходе непрерывного протекания болезни у них развиваются стереотипные движения рук, нерегулярное дыхание, атаксия и судороги.
После краткого периода псевдостабилизации, обычно в дошкольном или ранним школьном возрасте, состояние пациентов вновь ухудшается, появляется выраженная умственная отсталость, прогрессирующая спастичность, ригидность и сколиоз. Больные обычно доживают до взрослого возраста, однако продолжительность жизни уменьшена из-за повышения встречаемости необъяснимой внезапной смерти.
Кроме синдрома Ретта, мутации в гене МЕСР2 вызывают широкий спектр болезней, поражающих как мальчиков, так и девочек. Среди девочек симптоматика колеблется от сильно пораженных пациентов, не способных говорить, поворачиваться, сидеть или ходить, имеющих выраженную эпилепсию, до слабо пораженных пациентов, которые говорят, имеют сохранные двигательные функции, а также сравнительно хорошо сохранившуюся функцию рук.
У мальчиков колебания симптоматики — от внутриутробной гибели, врожденной энцефалопатии до умственной отсталости с различными неврологическими симптомами, или изолированной легкой умственной задержки; классический синдром Ретта описан только у мальчиков с соматическим мозаицизмом по мутации МЕСР2 или с дополнительной Х-хромосомой.
Лечение синдрома Ретта
Заподозренный на основе клинических признаков, диагноз синдрома Ретта обычно подтверждается ДНК-тестированием; тем не менее в настоящее время такое тестирование обнаруживает мутации в гене МЕСР2 только у 80-90% пациентов с типичным синдромом Ретта.
Клинические критерии диагноза для типичного синдрома Ретта включают нормальный пренатальный и перинатальный период, нормальную окружность головы при рождении, сравнительно нормальное развитие до 6-месячного возраста, задержку роста в возрасте между 6 и 48 мес, утрату приобретенных способностей и целенаправленных движений руками к 5-30 мес жизни, и последующее развитие стереотипных движений руками, потерю речевых навыков, выраженную психомоторную отсталость и развитие апраксическои походки и атаксии в возрасте между 12 и 48 мес жизни.
К настоящему времени эффективного лечения синдрома Ретта нет, и помощь сосредоточена на уходе и симптоматическом лечении. Медицинская помощь включает антиконвульсанты при судорогах, прием ингибиторов серотонина, карбидопы или леводопы при ригидности и мелатонина для улучшения сна. Семьи часто нуждаются в социальной поддержке, во взаимодействии с аналогичными семьями через группы взаимопомощи, а в некоторых случаях и в профессиональном консультировании.
Риски наследования синдрома Ретта
Приблизительно 99% случаев синдрома Ретта спорадические; большинство мутаций МЕСР2 возникают вновь, хотя в редких случаях они могут наследоваться от здоровой или мало пораженной матери со смещенной инактивацией Х-хромосомы. По крайней мере, 70% новых мутаций возникают в половых клетках отцов.
Если пара имеет больного ребенка, но мутация в гене МЕСР2 у родителей не выявлена, риск для будущих детей низкий, хотя и выше, чем в общей популяции, из-за возможности необнаруженного полового мозаицизма. Если же мать несет мутацию гена МЕСР2, каждый ребенок, независимо от пола, имеет 50% риск унаследовать мутацию.
Тем не менее недостаточная корреляция между генотипом и фенотипом у пациентов с мутациями в гене МЕСР2 обычно не позволяет давать прогнозы, разовьется ли у женского плода с мутацией МЕСР2 классический синдром Ретта или другая патология. Аналогично, идентификация мутации МЕСР2 у плода мужского пола также не позволяет предсказать внутриутробную гибель, развитие врожденной энцефалопатии или другой патологии.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Название протокола: Синдром Ретта
Синдром Ретта – прогрессирующее дегенеративное заболевание ЦНС, до нaстоящего времени обнaруживaемое только у девочек, при котором явно нормaльное рaннее рaзвитие осложняется чaстичной или полной утрaтой речи, локомоторных нaвыков и нaвыков пользовaния рукaми одновременно с зaмедлением ростa головы. [1,8]
Код протокола:
Код МКБ-10:
F84.2 Синдром Реттa
Сокращения, используемые в протоколе:
| АЛТ – аланинаминотрансфераза АСТ – аспартатаминотрансфераза ВВК – военно-врачебная комиссия В\м – внутримышечно В\в – внутривенно КТ – компьютерная томография ЛС – лекарственные средства МЗСР – Министерство здравоохранения и социального развития МНН – международное не патентованное название (генерическое название) МРТ – магнитно-резонансная томография МСЭК – медико-социальная экспертная комиссия ОАК – общий анализ крови ОАМ – общий анализ мочи ПЭТ – позитронно-эмиссионная томография РЭГ – реоэнцефалография РК – Республика Казахстан Р-р – раствор СИОЗС – селективные ингибиторы обратного захвата серотонина СПЭК – судебно-психиатрическая экспертная комиссия ЭКГ – электрокардиограмма ЭПО – экспериментально-психологическое обследование ЭЭГ – электроэнцефалограмма ЭхоЭГ – эхоэлектроэнцефалограмма |
Дата разработки протокола: 2015 год.
Категория пациентов: дети.
Пользователи протокола: детские психиатры.
Оценка на степень доказательности приводимых рекомендаций.
Шкала уровня доказательности:
| А | Высококачественный мета-анализ, систематический обзор РКИ или крупное РКИ с очень низкой вероятностью (++) систематической ошибки результаты. |
| В | Высококачественный (++) систематический обзор когортных или исследований случай-контроль или высококачественное (++) когортное или исследований случай-контроль с очень низким риском систематической ошибки или РКИ с не высоким (+) риском систематической ошибки. |
| С | Когортное или исследование случай-контроль или контролируемое исследование без рандомизации с не высоким риском систематической ошибки (+). Результаты, которых могут быть распространены на соответствующую популяцию или РКИ с очень низким или невысоким риском систематической ошибки (++ или +), результаты которых не могут быть непосредственно распространены на соответствующую популяцию. |
| D | Описание серии случаев или неконтролируемое исследование или мнение экспертов. |
| GPP | Наилучшая фармацевтическая практика. |
Классификация
Клиническая картина
Cимптомы, течение
Жалобы и анамнез:
Жалобы: отставание в психоречевом развитии, неадекватное поведение
Анамнез:
Пренатальный и перинатальный периоды без видимой патологии и психомоторное развитие в первые 5 месяцев без видимой патологии и нормальная окружность головы при рождении.
Задержка роста головы между 5 месяцами и 4 годами и потеря приобретенных целенаправленных ручных навыков в возрасте от 5 до 30 месяцев, что связано с одновременной дисфункцией общения и нарушенным социальным взаимодействием и появлением плохой координации (нестабильности) походки и (или) движений туловища.
Развитие тяжелых нарушений экспрессивной и рецептивной речи с выраженной психомоторной задержкой.
Стереотипные движения руками (такие как сжатие или вытирание рук) одновременно с потерей целенаправленных движений руками или после этого.
Дополнительные:
· дыхательные расстройства (периодическое апное во время бодрствования, перемежающееся гипервентиляцией, аэрофагия);
· судорожные припадки;
· спастичность, часто сочетающаяся с дистонией и атрофией мышц;
· сколиоз;
· задержка роста;
· гипотрофичные маленькие ступни.
Физикальное обследование: диагностически значимых изменений со стороны кожных покровов и внутренних органов (включая центральной и периферическую нервную систему) нет.
Диагностика
Диагностические исследования:
Основные (обязательные) диагностические обследования, проводимые на амбулаторном уровне:
· экспериментально-психологическое обследование.
Дополнительные диагностические обследования, проводимые на амбулаторном уровне:
· ОАК;
· ОАМ;
· биохимический анализ крови (печеночные пробы);
· ЭКГ – проводятся с целью мониторинга изменений соматического состояния на фоне основной терапии;
· ЭЭГ – при эпилептических и эпилептиформных пароксизмах.
Минимальный перечень обследований, которые необходимо провести при направлении на плановую госпитализацию, согласно внутреннему регламенту стационара с учетом действующего приказа уполномоченного органа в области здравоохранения.
Основные (обязательные) диагностические обследования, проводимые в стационаре:
· ОАМ – не реже 1 раза в месяц
· ОАК – не реже 1 раза в месяц
· биохимический анализ крови (АЛТ, АСТ, билирубин (прямой и непрямой, уровень глюкозы в крови) – не реже 1 раза в месяц;
· ЭКГ – не реже 1 раза в месяц;
· ЭПО (для поступивших впервые в жизни или впервые в текущем году). ЭПО для иных категорий пациентов – по решению лечащего врача.
Дополнительные диагностические обследования, проводимые в стационаре:
· ЭЭГ – при эпилептических и эпилептиформных пароксизмах.
Инструментальное обследование:
· ЭЭГ аномалии (медленный фоновый ритм и периодическое замедление ритма до 3-5 Гц, описаны центральновисочные Spikes, как при фрагильной Х-хромосоме и роландической эпилепсии).
Показания для консультации специалистов:
· консультация терапевта (педиатра) – исключение соматических заболеваний;
· консультация невропатолога – исключение текущих неврологических расстройств;
· консультация гинеколога (для женщин) – исключение гинекологических расстройств;
· консультации иных узких специалистов – сопутствующие соматические заболевания и\или патологические состояния.
Лабораторная диагностика
Дифференциальный диагноз
Таблица 1 – Дифференциальная диагностика:
| Параметры | Синдром Ретта | Аутизм |
| Клиническая картина | Нарастание когнитивного дефицита, признаки прогредиентности, присоединение неврологических сипмтомов | Признаки ИСКАЖЕННОГО психического развития, отсутствие прогредиентности. |
| Продолжительность | Не информативно | |
| Инструментальное обследование | Не информативно | |
Лечение
Цели лечения:
· устранение психопатологических нарушений;
· достижение медикаментозной ремиссии;
· стабилизация состояния пациента.
Тактика лечения:
При оценке микросоциальных условий как удовлетворительных и\или нетяжелом уровне расстройства рекомендуется преимущественно амбулаторное лечение. В случае усложненной клинической картины (за счет коморбидных состояний) и\или неэффективности вмешательства на амбулаторном этапе решается вопрос о госпитализации.
Немедикаментозное лечение:
Комплаенс - терапия, различные виды психотерапии, трудотерапия.
Режимы наблюдения (в зависимости от состояния пациента):
общий режим наблюдения – круглосуточное наблюдение без ограничения передвижения в отделении.
· режим частичной госпитализации – возможность нахождения в отделении в дневное или ночное время с учетом необходимости его адаптации во внебольничных условиях.
· режим лечебных отпусков – возможность нахождения, по решению ВКК вне отделения от нескольких часов до нескольких суток, с целью постепенной адаптации к внебольничным условиям, решения бытовых и социальных вопросов, а также оценки достигнутого лечебного эффекта.
· усиленный режим наблюдения – круглосуточное наблюдение и ограничение передвижения за пределами отделения.
· строгий режим наблюдения – круглосуточное непрерывное наблюдение, постоянное сопровождение медицинским персоналом в отделении и за его пределами.
Медикаментозное лечение:
Основные ЛС – препаратов, имеющих 100% вероятность применения нет.
Дополнительные ЛС(таблица 2 и 3):
Нейролептические препараты– предназначены для стабилизации психического состояния, устранения психопатологических расстройств и купирования двигательного возбуждения. Спектр препаратов невелик из-за возрастных ограничений (клозапин. галоперидол, трифлуоперазин,левомепромазин, хлорпромазин). Отдельную подгруппу составляют нейролептики пролонгированного действия, предназначенные для медикаментозного контроля психического состояния в амбулаторных условиях (галоперидол-деканоат,флуфеназин). Назначение атипичных нейролептиковпоследнего поколения возможно в случае доказанной (на заседании ВКК) неэффективности стандартной терапии и с письменного согласия законных представителей ребенка.
М-Н-Холинолитики – тригексифенидил–предназначен для коррекции побочных экстрапирамидных симптомов, вероятных на фоне терапии нейролептиками.
Антидепрессанты – предназначены для купирования сопутствующих депрессивных проявлений (амитриптилин,сертралин, флувоксамин).
Малые нейролептики – предназначены для коррекции поведенческих нарушений (хлорпротиксен,тиоридазин).
Транквилизаторы – предназначены для устранения тревожных расстройств и купирования двигательного возбуждения (диазепам,клоназепам).
Нормотимические (антиэпилептические) препараты – предназначены для стабилизации настроения, медикаментозного контроля нарушений биологических ритмов (карбамазепин, вальпроевая кислота, ламотриджин).
Медикаментозное лечение, оказываемое на амбулаторном уровне [4-7,9,12-18]:
Таблица 2 – Дополнительные медикаменты:
Рекомендуется монотерапия: одно из нижеперечисленных препаратов.
| МНН | Терапевтический диапазон | Курс лечения |
| Клозапин (УД – А) | 12,5- 100 мг\сутки внутрь | От нескольких месяцев до нескольких лет- до полной стабилизации поведения и психического состояния |
| Галоперидол (УД – А) | 2,5- 15 мг\сутки внутрь | |
| Трифлуоперазин (УД – А) | 2,5- 15 мг\сутки внутрь | |
| Левомепромазин (УД – В) | 12,5- 100 мг\сутки внутрь | |
| Галоперидол – деканоат (УД – А) | 25-100 мг\ в 4 недели в\м | |
| Флуфеназин (УД – А) | 12,5-50 мг\ в 4 недели в\м | |
| Амитриптилин (УД – А) | До 75мг\сутки внутрь | До исчезновения депрессивных проявлений |
| Сертралин (УД – А) | До 100мг\сутки внутрь | |
| Флувоксамин (УД – А) | 25-100мг\сутки внутрь | |
| Хлорпротиксен (УД – В) | До 100 мг\сутки внутрь | До исчезновения поведенческих нарушений |
| Тиоридазин (УД – В) | До 100мг\сутки внутрь | |
| Карбамазепин (УД – В) | До 400 мг\сутки внутрь | До признаков стабилизации эмоционального фона |
| Вальпроевая кислота (УД – В) | 300-600 мг\сутки внутрь | |
| Ламотриджин (УД – А) | До 100 мг\сут внутрь | |
| Карбамазепин (УД – В) | До 1г\сутки внутрь | До купирования эпиприпадков |
| Вальпроевая кислота (УД – В) | До 30мг\кг\сутки внутрь | |
| Ламотриджин (УД – А) | До 15мг\кг\сутки внутрь | |
| Тригексифенидил (УД – В) | 2-6мг\сутки внутрь | До исчезновения экстрапирамидных симптомов |
Медикаментозное лечение, оказываемое на стационарном уровне [4-7,9,12-18]:
Таблица 3 –Дополнительные медикаменты:
Рекомендуется монотерапия: одно из нижеперечисленных препаратов.
| МНН | Терапевтический диапазон | Курс лечения |
| Клозапин (УД – А) | 50-200 мг\сутки |
| МНН | Терапевтический диапазон |
| Левомепромазин(УД – В) | до 50 мг\сутки в\м |
| Хлорпромазин (УД – В) | до 50 мг\сутки в\м |
| Диазепам (УД – А) | 10-20 мг\сутки в\м |
Другие виды лечения: нет.
Хирургическое лечение: нет.
Индикаторы эффективности лечения:
· Непсихотический уровень психопатологических расстройств.
· Настроенность больного и\или его семьи на продолжение лечения на амбулаторном этапе.
· Отсутствие негативной реакции на необходимость приема психотропных средств.
Препараты (действующие вещества), применяющиеся при лечении
| Амитриптилин (Amitriptyline) |
| Вальпроевая кислота (Valproic Acid) |
| Галоперидол (Haloperidol) |
| Диазепам (Diazepam) |
| Карбамазепин (Carbamazepine) |
| Клозапин (Clozapine) |
| Клоназепам (Clonazepam) |
| Ламотриджин (Lamotrigine) |
| Левомепромазин (Levomepromazina) |
| Сертралин (Sertraline) |
| Тиоридазин (Thioridazine) |
| Тригексифенидил (Trihexyphenidyl) |
| Трифлуоперазин (Trifluoperazine) |
| Флувоксамин (Fluvoxamine) |
| Флуфеназин (Fluphenazine) |
| Хлорпромазин (Chlorpromazine) |
| Хлорпротиксен (Chlorprothixene) |
Госпитализация
Показания для госпитализации[2,3]:
Добровольная (экстренная и плановая) госпитализация:
· психопатологические расстройства психотического и\или непсихотического уровня с десоциализирующими проявлениями, проявления которых не купируются в амбулаторных условиях или
· решение экспертных вопросов (МСЭК, ВВК, СПЭК).
Принудительная госпитализация без решения суда – наличие психопатологических расстройств и действий, обусловливающих:
· непосредственную опасность для себя и окружающих;
· беспомощность, то есть неспособность самостоятельно удовлетворять основные жизненные потребности, при отсутствии надлежащего ухода;
· существенный вред здоровью вследствие ухудшения психического состояния, если лицо будет оставлено без психиатрической помощи.
Принудительная госпитализация – по определению суда, постановлению следственных органов и\или прокураторы.
Профилактика
Дальнейшее ведение (после стационара) – формирование и укрепление комплаенса.
Информация
Источники и литература
Информация
Конфликта интересов нет.
Условия пересмотра протокола: пересмотр протокола через 3 года после его опубликования и с даты его вступления в действие или при наличии новых методов с уровнем доказательности.

Синдром Ретта – прогрессирующее дегенеративное заболевание центральной нервной системы, названное по имени ученого, который впервые описал эту патологию.
Диагностируется синдром в течение первых 2-х лет жизни ребенка, причем возникает он обычно у детей при нормально протекающей беременности, родах и полноценном развитии в первые месяцы жизни (иногда до 1,5 лет). Затем происходит остановка развития и регресс всех форм психической деятельности, что сопровождается возникновением аутизма, моторной стереотипии, прогрессирующего моторного снижения. В последующем это заболевание приводит к инвалидности и даже к смерти.
Основными проявлениями болезни являются обратное развитие уже приобретенных двигательных и речевых навыков в возрасте от 1,5 до 3-4 лет, повторяющиеся стереотипные и неконтролируемые движения рук, умственная отсталость.

Что это такое?
Синдром Ретта — психоневрологическое наследственное заболевание, встречается почти исключительно у девочек с частотой 1:10000 — 1:15000, является причиной тяжёлой умственной отсталости у девочек.
Впервые болезнь была описана австрийским неврологом Андреасом Реттом (нем. Andreas Rett) в 1966 году. Развитие ребёнка до 6 — 18 месяцев протекает нормально, но потом у девочки начинают пропадать приобретённые речевые, двигательные и предметно-ролевые навыки.
Характерным для данного состояния являются стереотипные, однообразные движения рук, их потирание, заламывание, при этом не носящие целенаправленного характера. Речь затрудняется, ответы становятся однообразными или эхолалическими, временами речь совсем пропадает (мутизм).
Причины возникновения
В 90-х годах существовала гипотеза о том, что синдром Ретта – это определённое нарушение, которое связано с генными мутациями, локализующимися в Х-хромосоме; обусловленное доминантным признаком и у мальчиков не может совмещаться с жизнью. В дальнейшем подтвердилась передача гена-мутанта Х-хромосомой отца тем, что это наследственная патология очень редко может встретиться у мальчиков, так как они получают от отца Y-хромосому. Именно поэтому при семейном типе наследования синдрома Ретта, мальчики в таких семьях рождаются практически здоровыми.
В настоящее время существуют подтверждения о наследственной природе заболевания. Генетическую причину возникновения синдрома Ретта связывают с изменённой Х-хромосомой и мутациями, которые происходят в генах, регулирующих процесс репликации. В данном случае происходит дефицит некоторых белков, которые регулируют этот рост, а также нарушается холинергическая их функция.
Для синдрома Ретта была выдвинута гипотеза о прерванном развитии, для которого характерен дефицит нейротрофических факторов. Таким образом, поражаются базальные ганглии, нижние моторные нейроны, вовлекается спинной мозг и гипоталамус. Анализируя морфологические изменения, учёные пришли к выводу, что происходят замедления в развитии мозга с самого рождения, который полностью останавливается в росте к четырём годам. А также у таких детей отмечается замедление в росте тела и некоторых соматических органов.
Стадии развития
Синдром Ретта формируется в течение длительного времени, поэтому заболевание принято подразделять на несколько стадий в зависимости от ухудшения состояния больного:
- Стагнация — временное приостановление болезни, при которой не происходит нарастания симптомов. Длится от 6-18 месяцев и более. Ребенок теряет интерес к окружающим событиям, заметна гипотония мышц, замедление роста головы и конечностей.
- Ухудшение состояния. Стадия длится от 1 года до 3-4 лет. Если ребенок овладел навыками речи, передвижения, они постепенно исчезают. Появляются стереотипные манипуляции руками, нарушения со стороны легочной системы (гипервентилляция, одышка, внезапная остановка дыхания), дискоординация движений, немотивированное беспокойство. Уже на 2 стадии появляются судорожные припадки, лечение которых не является результативным.
- Относительная стабильность, эта стадия может продолжаться вплоть до раннего школьного возраста. Отмечается умственная недостаточность, судороги, малая прибавка веса, нарушение эмоционального контакта с окружающими. Эпилептические припадки сменяются заторможенностью нервной системы.
- Завершающая стадия характеризуется снижением частоты судорог, зато появляется кахексия, сколиоз, выраженные нарушения со стороны дыхания. Возможна несостоятельность передвижения, определяют низкий рост конечностей и малую окружность головы.
Характерные симптомы, присущие той или иной стадии, не являются на сто процентов условными и могут варьироваться в зависимости от скоротечности болезни и индивидуальных клинических случаев.

Симптомы синдрома Ретта
Отдельно следует акцентировать внимание на основных симптомах, по которым определяется синдром Ретта, поскольку в медицинской практике были известны случаи, когда из-за неверной трактовки признаков болезни ставился совершенно другой диагноз, что приводило в итоге к быстрому летальному исходу.
Синдром Ретта определяется по таким критериям:
- Ярко выраженная микроцефалия. В период после рождения у ребенка наблюдается нормальное соотношение размера головы с туловищем. Постепенно рост головы замедляется, что вызвано уменьшением размеров головного мозга.
- Развивающийся сколиоз. Нарушения позвоночного отдела характерны для всех детей, страдающих данным недугом. Причиной искривления спины является дистония мышц.
- Ментальное развитие. Синдром Ретта характеризуется глубокой умственной отсталостью и отсутствием насыщенной познавательной деятельности, которая обычно присутствует у всех маленьких детей. Многие пациенты поначалу приобретают навыки говорения и восприятия окружающего мира, однако со временем полностью их теряют. У ребенка заметно отсутствие любого экспрессивного или импрессивного общения с окружением. Для определения нарушений в ментальном развитии специалисты используют стандартизированные психологические тесты.
- Специфические движения рук. У детей пропадают навыки держания каких-либо предметов, будь то игрушка или бутылочка с молоком. При этом возникают монотонные движения рук, напоминающие мытье под краном, а также характеризующиеся сжиманием, перебиранием пальцев, хлопками на уровне груди, лица и за спиной. Также пациент может сосать или покусывать руки, хаотично ударять себя ими по разным частям тела.
- Судорожные припадки. Практически в 80% случаев девочки страдают от эпилептических припадков, которые также сопровождаются парциальными приступами, дроп-атаками, тонико-клоническим типом припадка. Синдром Ретта характеризуется такими признаками формирования предприпадочного состояния: тремор, затрудненность дыхания, резкие движения, направленность взгляда в одну точку с полным при этом онемением тела. Данные симптомы нередко лечатся антиконвульсантами, однако подобные препараты не дают никакого положительного эффекта, поскольку перечисленные признаки не относятся к группе судорожных заболеваний, а представляют собой лишь синдром Ретта.
- Нейрохимические симптомы. Во время исследования пациентов, скончавшихся на тяжелой стадии синдрома Ретта, ученые определили, что размеры их мозга были меньше нормы на 12% или 24%, в зависимости от возрастной группы, в которой находился пациент. В мозжечке, коре полушарий головного мозга и спинальных ганглиях наблюдался дефицит нейронов и глиоз, а также пониженный уровень пигментации. Согласно морфологическим исследованиям к четырем годам у людей, страдающих синдромом Ретта, диагностировалась полная остановка развития мозга и замедление роста других органов и частей тела.
Диагностика
Консультация психиатра начинается со сбора анамнеза. Во время беседы с родителями специалист выясняет:
- нормально ли протекали беременность и роды;
- как развивался ребёнок на протяжении первых 6–18 месяцев;
- какова динамика роста головы;
- когда ребёнок начал терять приобретённые навыки;
- имеются ли у пациента стереотипные движения рук, судорожные припадки, нарушения речи, дыхания, походки и координации, задержка психомоторного развития.
Для точной диагностики врач часто назначает дополнительные обследования:
- ЭЭГ (электроэнцефалограмма), измеряющая биоэлектрическую активность головного мозга (медленный фоновый ритм — свидетельство мутации в Х-хромосоме);
- КТ головного мозга, позволяющее обнаружить изменения, указывающие на прекращение развития мозга;
- УЗИ внутренних органов, дающее понятие о степени их развития.
При отсутствии данных обследований синдром Ретта можно спутать с аутизмом. Для обоих этих состояний характерны:
- снижение способности к обучению;
- потеря речи;
- отсутствие контроля за тазовыми органами;
- уход от внешнего мира;
- отсутствие зрительного контакта;
- нежелание идти на эмоциональный и социальный контакт;
- нарушение чувствительности тела;
- беспричинные крики и плач.
Отличить одно заболевание от другого можно с помощью дифференциальной диагностики, разработанной в 1988 году на Международной конференции по синдрому Ретта.
Данные дифференциальной медицинской диагностики:
| Описание симптома | Проявление при синдроме Ретта | Проявление при раннем аутизме |
| Замедленный рост кистей рук, стоп и головы | Характерный признак синдрома Ретта | Признак отсутствует |
| Отставание в развитии в возрасте от шести месяцев до года | Не проявляется | Часто наблюдается |
| Дыхательные расстройства | Часто наблюдаются | Не проявляются |
| Стереотипные движения рук | Повторяющиеся однообразные движения рук в области пояса | Разнообразные и сложные движения, не ограниченные зоной пояса |
| Эпилептические припадки | Часто повторяются | Редко проявляются |
| Координация движений | Прогрессирует нарушение координации, переходящее в полную неподвижность | Движения и походка почти нормальные, но кажутся манерными |
Используя дифференциальную диагностику, отличить аутизм от синдрома Ретта могут не только врачи, но и сами родители.
Лечение
На данном этапе развития медицины синдром Ретта является неизлечимым заболеванием. Однако с помощью лекарственных препаратов, реабилитационных методик и специальной диеты можно добиться улучшения состояния ребёнка, предупредить серьёзные деформации тела и улучшить качество жизни больного.
Для улучшения общего состояния и смягчения симптомов врач может прописать следующие медикаменты:
- противопаркинсонические препараты (Бромокриптин, Перлодел);
- ноотропы для улучшения работы мозга (Кортексин, Церебролизин, Цераксон);
- лекарства для регулирования биологического режима дня и ночи (Мелатонин);
- препараты для успокоения нервной системы и коррекции поведения (Ноофен, Фенибут, Глицин);
- противоэпилептические препараты для уменьшения количества приступов (Карбамазепин, Ламотриджин);
- средства для поддержки работы внутренних органов: сердца, печени, желудка, кишечника, поджелудочной железы.

Реабилитация
В программу реабилитации может входить:
- Консультации психолога и логопеда — один раз в неделю.
- Музыкотерапия — повышает коммуникативную активность ребёнка.
- Массаж и лечебно-оздоровительная физкультура — направлены на укрепление мышц, увеличение их тонуса.
Хорошие отзывы имеет метод, придуманный французским отоларингологом Альфредом Томатисом. Целью является повторное обучение ребёнка процессу слушания, что улучшает способности к изучению и освоению языков, увеличивает творческий потенциал и положительно влияет на социальное поведение малыша.
- Арт-терапия и дельфинотерапия — положительно воздействуют на психоэмоциональное состояние ребёнка.
- Иппотерапия (лечебная верховая езда) и гидротерапия (душ, обливания, обтирания) — оказывают биомеханическое воздействие на тело ребёнка, укрепляет мышцы.
- АВА-терапия — улучшает социальную адаптацию больных. Какие-то сложные действия (контактность, творческая игра, речь) для ребёнка разбиваются на мелкие части. В будущем они соединяются в один большой блок.
Стоит отметить, что хорошее терапевтическое действие оказывает посещение остеопата. Улучшение состояния наблюдается уже после нескольких сеансов.
Особенности питания
Питание больного ребёнка представляет определённые сложности. У многих девочек наблюдается повышенное слюнотечение, плохое состояние ротовой полости, поэтому накормить их настоящая проблема. Некоторые дети имеют хороший аппетит и с удовольствием употребляют любимые продукты. Но все они едят очень медленно, процесс приёма пищи может растягиваться до полутора часов. Что касается питья, то почти у половины больных малышей есть трудности с глотанием, которые проявляются поперхиванием, кашлем и могут грозить попаданием жидкости в дыхательные пути.
- Детям с синдромом Ретта тяжело пережёвывать пищу, содержащую грубые волокна (мясо, сырые овощи, фрукты), поэтому её нужно измельчать и давать в виде пюре. Гарниры лучше предлагать мелкими кусочками или в размятом виде. Во время кормления нужно следить, чтобы голова ребёнка была под правильным углом, не запрокидывалась.
- В некоторых случаях, когда процесс поглощения пищи становится слишком проблематичным и болезненным, имеет смысл кормить ребёнка через зонд специальными питательными смесями. Такой вариант позволяет значительно улучшить качество жизни малыша и может стать настоящим для него спасением.
Многие дети страдают от тошноты и часто отказываются от еды, поэтому накормить их бывает очень трудно, такие малыши быстро теряют в весе. Поэтому пища должна быть обогащённой белками и жирами, достаточно калорийной и витаминизированной. Рекомендуется кормить ребёнка малыми порциями и часто (каждые 3 часа), чтобы не перегружать пищеварительную систему. Детей младшего возраста поят витаминизированным молоком или молочными смесями.
Прогноз
Ведется активная разработка стратегии использования стволовых клеток, на которых будет основано лечение синдрома Ретта. Уже сегодня предварительные средства опробованы на лабораторных мышах. Опыты профессора Беличенко, которые проводились в Калифорнийском университете, дают положительный прогноз и надежду на скорое научное открытие эффективного лечения синдрома Ретта.
Профилактика
Лечебная физкультура — один из оптимальных способов коррекции двигательных расстройств. Она включает упражнения, направленные на поддержание гибкости и амплитуды движений конечностей, а также как можно более длительное сохранение навыка ходьбы.
Предлагаются психологические программы максимального развития оставшихся сохранными двигательных навыков и формирования на их основе “языка общения”. Используется также музыкальная терапия, так как она оказывает благоприятный успокаивающий эффект на детей и частично компенсирует нарушение контакта с окружающим миром. Исследования синдрома Ретта интенсивно ведутся во всем мире, и открытие его специфического биологического маркера является, вероятно, лишь вопросом времени.
Когда это произойдет, возникнут новые перспективы лечения патологии или облегчения состояния больных, а также возможности пренатального скрининга и предотвращения этого тяжелого заболевания.
Читайте также: