
Синдром дисомии по y хромосоме кратко
Обновлено: 04.07.2024
47, XYY синдром является анеуплоидией половой хромосомы, при которой мальчики имеют дополнительную Y хромосому.
Распространенность 1-5 / 10 000.
Взрослые носители кариотипа 47,XYY в большей части случаев имеют нормальный мужской фенотип. Добавочная (отцовская) Y-хромосома появляется чаще всего в результате нерасхождения хроматид во 2-м делении мейоза. Возраст отца не является фактором риска.
Синдром характеризуется клинически: высоким ростом с самого детства, макроцефалией, характерными чертами лица (мягкий гипертелоризм, низкопосаженные уши, легкое уплощение скуловой области), задержкой речи и повышенным риском возникновения социальных и эмоциональных нарушений, синдромом дефицита внимания с гиперактивностью, аутизмом.
Иногда наблюдаются изменения ЭКГ, шаровидные или абсцедирующие угри и варикозное расширение вен, однако повышенный риск возникновения этих расстройств у лиц с кариотипом 47,XYY не подтвержден. Умственное развитие в пределах нормы, но речевое развитие задерживается. Нередко подростки и мужчины с кариотипом 47,XYY очень агрессивны, склонны к преступным действиям и плохо адаптируются к жизни в обществе. У большинства развитие и функции половых желез нормальные, однако известны случаи недоразвития яичек, бесплодия или пониженной фертильности.
Лечение не требуется. Если кариотип 47,XYY обнаружен в ходе пренатального исследования или у ребенка в препубертатном периоде, нужно правдиво и подробно проконсультировать родителей. Взрослый мужчина, у которого впервые выявлен кариотип 47,XYY, нуждается в психологической поддержке; могут потребоваться медико-генетические консультации.
Супружеским парам, в которых мужчина несет кариотип 47,XYY, рекомендуют провести пренатальную диагностику, хотя в таких семьях дети обычно имеют нормальный кариотип.
Кравец В.С. 1, 2, 3 Ворсанова С.Г. 1, 2, 3 Юров И.Ю. 2, 1, 4 Колотий А.Д. 1, 2 Боченков С.В. 1 Гордеева М.Л. 1 Юров Ю.Б. 1, 2, 3
В статье рассматривается редкий случай задержки психоречевого развития, признаков аутизма и микроаномалий у мальчика 3 лет с дисомией хромосомы Y, рождённого в кровнородственном браке. Отмечается, что клинические признаки, обнаруженные у пробанда, не совсем совпадают с классическими симптомами синдрома дисомии хромосомы Y. Подчеркивается необходимость применения молекулярно-цитогенетических технологий, таких как флюоресцентная in situ гибридизация (FISH) и молекулярное кариотипирование (серийная сравнительная геномная гибридизация или arrayCGH) с целью выявления возможных геномных аномалий, не обнаруживаемых стандартным (классическим) цитогенетическим методом, способных повлиять на клиническую картину, отмеченную у данного пациента. Обсуждается возможное влияние на симптомокомплекс пробанда сочетания дисомии хромосомы Y и инбридинга (коэффициент инбридинга 1/32) в данной семье.

1. Ворсанова С.Г., Шаронин В.О., Курило Л.Ф. Аномалии половых хромосом при нарушении репродуктивной функции у мужчин : обзор литературы // Пробл. репродукции. – 1998. – № 2. – С. 12-21.
3. Ворсанова С.Г., Воинова В.Ю., Юров И.Ю., Куринная О.С., Демидова И.А., Юров Ю.Б. Цитогенетические, молекулярно-цитогенетические и клинико-генеалогические исследования матерей детей с аутизмом: поиск семейных генетических маркеров аутистических расстройств // Журнал неврологии и психиатрии им. C.С. Kорсакова. - 2009. - Т. 109, № 6. - С. 54-64.
4. Ворсанова С.Г., Юров И.Ю., Куринная О.С., Воинова В.Ю., Юров Ю.Б. Геномные аномалии у детей с умственной отсталостью и аутизмом: использование технологии сравнительной геномной гибридизации на хромосомах in situ (HR CGH) и молекулярного кариотипирования на ДНК-микроматрицах (arrayCGH) // Журнал неврологии и психиатрии им. C.С. Корсакова. - 2013. - Т. 113, № 8. - С. 46-49.
5. Ворсанова С.Г., Юров И.Ю., Демидова И.А., Кравец В.С., Юров Ю.Б. Цитогенетика и молекулярная цитогенетика аутизма. - М. : Издательский дом Академии Естествознания, 2016. – 144 с.
7. Юров И.Ю., Ворсанова С.Г., Юров Ю.Б. Cовременные достижения в молекулярно-цитогенетической диагностике наследственных болезней (лекция) // Клиническая лабораторная диагностика. - 2005. - № 11. - С. 21-29.
8. Юров И.Ю., Ворсанова С.Г., Юров Ю.Б. Геномные и хромосомные болезни центральной нервной системы: молекулярные и цитогенетические аспекты. - М. : Медпрактика, 2014. – 384 с.
9. Юров И.Ю., Ворсанова С.Г., Зеленова М.А., Васин К.С., Юров Ю.Б. Биоинформатическая технология оценки функциональных последствий геномных вариаций // Фундаментальные исследования. – 2015. - № 2-19. – С. 4209-4214.
10. Юров Ю.Б., Ворсанова С.Г. Молекулярно-цитогенетические исследования хромосомных аномалий и нарушений при нервно-психических заболеваниях: поиск биологических маркеров для диагностики // Вестник РАМН. - 2001. - № 7. - С. 26-31.
11. Iourov I.Y., Vorsanova S.G., Yurov Y.B. Single cell genomics of the brain: focus on neuronal diversity and neuropsychiatric diseases // Current Genomics. - 2012. - Vol. 13. – N 6. - Р. 477-488.
12. Iourov I.Y., Vorsanova S.G., Korostelev S.A., Zelenova M.A., Yurov Y.B. Long contiguous stretches of homozygosity spanning shortly the imprinted loci are associated with intellectual disability, autism and/or epilepsy // Mol Cytogen. – 2015. – Vol. 8. – № 1. – 8 р.
13. Milazzo J.P., Rives N., Mousset-Simeon N., Mace B. Chromosome constitution and apoptosis of immature germ cells present in sperm of two 47,XYY infertile males // Hum. Reprod. – 2006. – 21. – P. 1749–1758.
14. Robinson D.O., Jacobs P.A. The origin of the extra Y chromosome in males with a 47, XYY karyotype // Hum. Mol. Genet. – 1999. – N 8. – P. 2205–2209.
15. Schinzel A. Catalogue of unbalanced chromosome aberrations in man. - Berlin, New York, de Gruyter, 2001. – 966 с.
16. Shah K., Sivapalan G., Gibbons N., Tempest H., Griffin D.K. The genetic basis of infertility // Reproduction. – 2003. – N 126. – P. 13-25.
17. Soloviev I.V., Yurov Y.B., Vorsanova S.G., Malet P. Microvawe acnivation of fluorescence in situ hybridization: a novel method for rapid chromosome detection and analysis // Focus. – 1994. – N 16 (4). – P. 115-116.
18. Soloviev I.V., Yurov Y.B., Vorsanova S.G., Fayet F., Roizes G., Malet P. Prenatal diagnosis of trisomy 21 using interphase fluorescence in situ hybridization of postreplicated cells with site-specific cosmid contig probes // Prenatal diagn. – 1995. – № 15. – Р. 237-238.
19. Yurov Y.B., Laurent A.-M., Marcais B., Vorsanova S.G., Roizes G. Analysis of pericentromeric chromosome 21 specific YAC clones by FISH: identification of new markers for molecular cytogenetic application // Hum. Genet. – 1995. – № 95. – Р. 287-293.
20. Yurov Y.B., Vorsanova S.G., Soloviev I.V., Demidova I.A., Alexandrov I.A., Sharonin V.O., Beresheva A.K. Original collection of DNA probes for preimplantational, fetal prenatal and postnatal diagnosis of chromosomal analysis by FISH / (eds): Macek M. Sr., Bianchi D., Cuckle H. Early prenatal diagnosis, fetal cells and DNA in mother, present state and perspectives. – Prague, 2002. - P. 275-283.
Среди обширного спектра хромосомных и геномных аномалий, обнаруживаемых у детей с задержкой психоречевого и полового развития, видное место занимают различные численные и структурные аномалии половых хромосом (гоносом). Наиболее изучены и часто встречаются синдромы Шерешевского-Тернера (кариотипы - 45,Х; 46,X,i(Xq); 45,X/46,XХ и другие), Клайнфельтера (кариотипы - 47,XXY; 48,XXXY и другие), трисомии хромосомы Х (кариотип - 47,ХХХ) и дисомии хромосомы Y (кариотип - 47,XYY) [2; 15]. Наблюдаются как регулярные, так и мозаичные формы этих синдромов; возможен тканевой мозаицизм. Клинические признаки пациентов сильно варьируют от почти полного их отсутствия, особенно в случаях мозаицизма с малой долей аномального клона, до выраженной умственной отсталости, пороков и микроаномалий развития, нарушения репродуктивных функций и других симптомов. Диагностика подобных случаев нередко, особенно при мозаицизме и структурных перестройках, требует применения таких молекулярно-цитогенетических методов исследований, как флюоресцентная гибридизация in situ (FISH) с хромосомоспецифичными и сайтспецифичными ДНК зондами [10], метафазная сравнительная геномная гибридизация, а также серийная сравнительная геномная гибридизация на ДНК-микроматрицах (array CGH), что позволяет уточнить генетический диагноз и проводить корректное медико-генетическое консультирование семей [3; 4; 7; 11].
Материалы и методы
В работе обследовался мальчик в возрасте 3 лет. Культура лимфоцитов периферической крови, приготовление препаратов, дифференциальное окрашивание хромосом по длине и анализ кариотипа проводились по стандартным методикам (GTG- и CBG-окрашивание хромосом по длине) [2]. Кроме классического цитогенетического анализа, также было проведено молекулярно-цитогенетическое исследование – флюоресцентная in situ гибридизация (FISH) с центромерными хромосомоспецифичными ДНК-зондами на хромосомы Х и Y из оригинальной коллекции лаборатории [10; 18; 20]. Гибридизация и анализ препаратов проводились по стандартным протоколам [17; 19].
Результаты
В клинику института для обследования поступил ребёнок 3 лет, родившийся от 2-й беременности, 2-х физиологических родов с массой тела 3750 г, длиной - 54 см. От 1-й беременности родился здоровый мальчик, ему 5 лет. Причиной обращения послужили следующие клинические признаки: задержка психоречевого развития и аномалии поведения. При осмотре ребёнка отмечены такие симптомы, как ограниченность понимания обращённой речи, несформированность навыков опрятности и самообслуживания, стереотипные действия с предметами неигрового назначения, резко отрицательная реакция на запреты и ограничения в виде падения на пол и ударов головой об пол, а также агрессии к окружающим (укусы и т.д.), ходьба на цыпочках. Кроме того, у пациента обнаружены хронические кататоно-аффективные расстройства, аллергические реакции на пыль, плесень и шоколад, дисфункция желчного тракта, плоско-вальгусная деформация стоп, пролапс митрального клапана. Из родословной (рис. 1) видно, что прапрадеды пробанда по отцовской и материнской линиям являются родными братьями, и таким образом, родители пробанда – троюродные сибсы. В родословной, со слов родителей, не выявлено случаев умственной отсталости или пороков развития, у деда по отцовской линии обнаружен поллиноз, у деда по материнской линии – ишемическая болезнь сердца (ИБС).
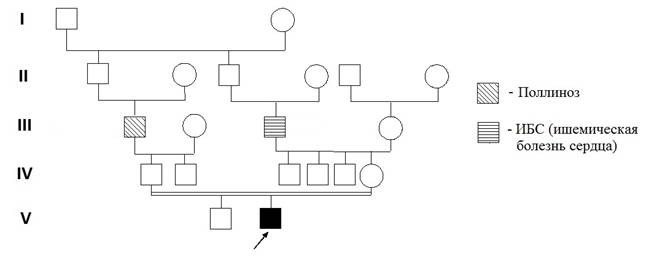
Рис. 1. Родословная пациента с синдромом дисомии хромосомы Y
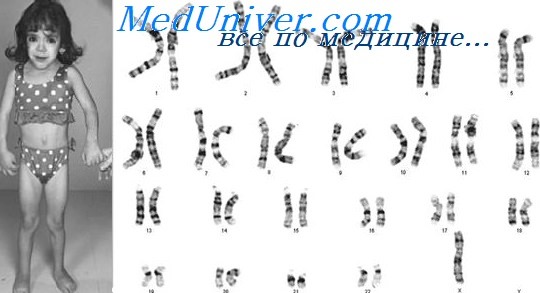
На основании клинических признаков принято решение о проведении цитогенетического исследования. Из анализа кариотипа (рис. 2) видно, что у пробанда выявлены две хромосомы Y. В результате проведённых цитогенетических исследований кариотип пробанда (GTG- и CBG-окрашивания хромосом по длине) - 47,XYY. Других численных или структурных аномалий хромосом, обнаружимых стандартными цитогенетическими методами, у данного пациента не выявлено.
Для исключения мозаицизма проведено FISH исследование (рис. 3), при котором не обнаружено предполагавшегося хромосомного мозаицизма гоносом, и дисомию хромосомы Y следует считать регулярной и возникшей de novo: 47,XYY. ish (DXZ1x1;DYZ3x2) [100]. nuc ish (DXZ1x1;DYZ3x2) [1000]. Таким образом, после проведения цитогенетического и молекулярно-цитогенетического исследований ребёнку поставлен диагноз – синдром дисомии хромосомы Y.
На основании результатов цитогенетических и молекулярно-цитогенетических исследований с учётом родства родителей принято решение о дополнительном проведении пробанду молекулярно-цитогенетического исследования – серийной сравнительной геномной гибридизации (молекулярного кариотипирования или array CGH) [4; 8; 11]. Проведение молекулярного кариотипирования данному пациенту позволит выявить возможные аномалии генома, а также установить участки потери гетерозиготности (loss of heterozygosity, LOH), которые в данном случае ввиду родства родителей (троюродные сибсы) могут быть обширны и способны значительно (негативно) повлиять на клинические проявления у пробанда [12].
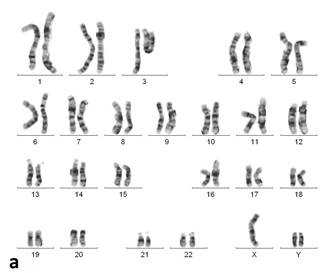

Рис. 2. Кариотип пробанда после проведения GTG- (а) и CBG- (б) окрашивания (47,XYY)
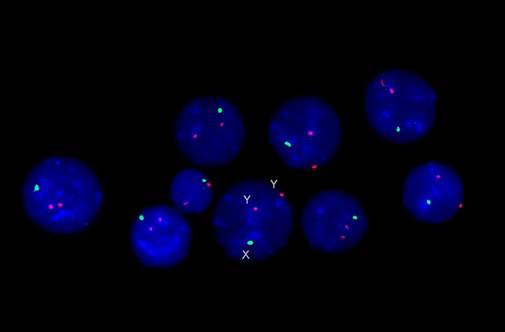
Рис. 3. Результаты FISH-исследования с центромерными ДНК-зондами на хромосомы Х и Y (зеленые сигналы – хромосома Х, красные – хромосома Y)
На рисунке представлены интерфазные ядра лимфоцитов периферической крови, в каждом из которых по две хромоcомы Y.
В достаточно широком спектре различных численных и структурных аномалий половых хромосом (гоносом) дисомия хромосомы Y занимает особое место благодаря исключительной вариабельности клинических признаков данного синдрома: от почти полного отсутствия каких-либо патологических проявлений до умственной отсталости тяжёлой степени; бесплодие наблюдается лишь в 30% пациентов, и прежде всего это связано с нарушением сперматогенеза. Причины клинического полиморфизма при этом синдроме не всегда ясны и могут быть связаны с иными хромосомными (геномными) микроаномалиями, не обнаружимыми стандартными методиками классического кариотипирования [4; 7].
Данный случай необычен тем, что у пациента с регулярной дисомией хромосомы Y отмечены выраженные черты аутизма, что при данной патологии наблюдается нечасто [4]; вероятно, на картину заболевания повлияло и то, что ребёнок рождён в кровнородственном браке (родители – троюродные сибсы и принадлежат к одному и тому же этносу (аварцы) Северного Кавказа, где кровнородственные браки у некоторых народов нередки); поэтому у пробанда возможно наличие обширных участков потери гетерозиготности (LOH) более чем в 3% генома (коэффициент инбридинга 1/32) [6; 12]. Именно молекулярное кариотипирование и биоинформатический анализ in silico позволит выявить данные участки, что при их наличии может привести и к возникновению различных наследственных аномалий [8; 9]. С учётом всех данных этой семье рекомендовано медико-генетическое консультирование с возможной пренатальной диагностикой при повторном деторождении.
Заключение
В данной статье мы рассматриваем необычный случай синдрома дисомии хромосомы Y у мальчика, рождённого в кровнородственном браке. Изучение влияния на клинические проявления у пробанда как регулярной формы дисомии хромосомы Y, так и кровнородственного брака весьма важно для понимания вариабельности клинических проявлений синдрома дисомии хромосомы Y. Проведение array CGH (молекулярного кариотипирования на ДНК-микроматрицах) с последующим биоинформатическим анализом позволит выявить возможные дополнительные микроаномалии генома, не обнаруженные классическими цитогенетическими методами, а также участки потери гетерозиготности (LOH), которые тоже могут влиять на фенотип [8; 9].
Исследование выполнено при финансовой поддержке Российского научного фонда (проект № 14-15-00411).
Анеуплоидии половых хромосом: XYY синдром, XXY синдром (синдром Клайнфельтера), Х0 синдром (45Х0 синдром, синдром Тернера), ХХХ синдром
В большинстве случаев анеуплоидий половых хромосом (встречающихся приблизительно у 0,3% живых новорожденных) отмечается повышенный риск психиатрических отклонений. С другой стороны, анеуплоидии половых хромосом относительно часто встречаются среди детей, состоящих на учете психиатра. По результатам одного из исследований (Crandall et al., 1972) 1,6% детей, обратившихся к детскому психиатру, имели анеуплоидии половых хромосом.
Данные аномалии связаны с несколько повышенным риском задержки умственного развития. Например, по результатам исследования Ratcliffe et al. (1994) было продемонстрировано, что кариотип XXY выявлялся у 5,4 на 1000 фенотипических мальчиков, наблюдавшихся в учреждениях для лиц с умственной отсталостью, а коэффициент IQ у таких детей был несколько ниже, чем в контрольной группе.
Средний коэффициент IQ у девочек с тремя Х-хромосомами составил 86,8, у мальчиков с кариотипом XXY — 91,4, а у мальчиков с кариотипом XYY — 101,7; все показатели были значимо ниже, чем в контрольных группах (109,1 —у девочек и 115,8 — у мальчиков). Среди детей с дополнительной половой хромосомой также отмечалась меньшая окружность головы (даже при рождении), что отражает воздействие дополнительных половых хромосом на рост головного мозга в пре- и постнатальном периоде. Кариотип XXX был выявлен у 4,3 из 1000 фенотипических девочек, находящихся в лечебных учреждениях (Pennington et al., 1980).
Средний коэффициент IQ при кариотипе ХО (синдром Тернера) находится в пределах нормы, но многие девочки с данным кариотипом путают право-лево и имеют отклонения организации восприятия, их вербальный 1Q с определенной индивидуальной вариабельностью имеет склонность превышать IQ исполнения заданий (Temple и Carney, 1993).
Более серьезная умственная отсталость, очевидно, присуща пациентам со сложными аномалиями, к примеру, фенотипическим мужчинам с тремя и более Х-хромосомами (Zaleski et al., 1966) или с дупликацией обеих половых (X и Y) хромосом (Schlegel et al., 1965). Тем не менее, основные отклонения, встречающиеся у таких детей, включают сложности обучения и восприятия и поведенческие отклонения (Bender et al., 1983, Ratcliffe et al., 1994).
а) XYY синдром. У мальчиков с дополнительной Y-хромосомой (около 0,1% всего мужского населения) имеется значительно повышенный риск задержки и других отклонений речевого развития, приступов гнева и агрессивности. У таких пациентов также часто отмечается гипотония и раннее появление дефицита внимания с гиперактивностью. Приступы гнева часто встречаются в раннем детском возрасте. Распространена сниженная общительность, а риск аутизма повышен (Hagerman, 1989). Показатель IQ обычно в пределах нормы, но очень часто отмечаются трудности в обучении, а частота задержки умственного развития по сравнению с общей мужской популяцией повышена.
На основании как минимум одного объективного проспективного исследования предполагается небольшое повышение склонности к агрессивному поведению и проявления садизма в половой жизни во взрослом возрасте (Schiavi et al., 1988). Большинство пациентов имеет высокий рост и пропорциональное строение относительно размеров ног и головы. Пациенты обычно не менее чем на 13 см выше своего отца. Обследование на выявление кариотипа XYY необходимо у высокорослых мальчиков с отклонениями в поведении и трудностями в обучении.
Синдром Клайнфельтера
б) XXY синдром (синдром Клайнефельтера). У мальчиков с одной или более дополнительной Х-хромосомой (около 0,1-0,2% всех новорожденных мальчиков) диагностируется синдром Клайнфелтера (Lanfranco et al., 2004). Почти 2/3 случаев составляет кариотип 47XXY. Окружность головы, рост и вес при рождении ниже средних параметров. Приблизительно с возраста 2-4 лет отмечается увеличение скорости роста, особенно длины ног. Средний рост пациентов во взрослом возрасте приблизительно на 13 см превышает рост отца. Размер головы не компенсируется.
Дополнительная Х-хромосома ингибирует рост головного мозга во внутриутробном периоде. У мальчиков с синдромом Клайнфелтера обычно отмечается снижение вербального IQ, в то время как общий коэффициент IQ остается в пределах нормы (или слегка снижен); обычно коэффициент IQ варьирует от 60 до 130. Отмечается задержка речевого развития, и в большинстве случаев лечение по поводу отклонений развития речи начинается задолго до проведения хромосомной диагностики. Пациенты часто бывают неуклюжими и имеют отклонения типичные для детей с DAMP (дефицит внимания (Deficits in Attention), двигательного контроля (Motor control) и восприятия (Perception), иногда с тенденцией к гипоактивности.
Данные отклонения могут усиливаться за счет аномального строения тела и нервно-мышечных характеристик. Нередки серьезные проблемы с чтением и письмом, не зависящие от общего IQ. Изменения на МРТ в наиболее пораженных областях (лобной, височной и моторной зонах) и менее затронутой теменной области соответствуют выявляемым когнитивным и поведенческим отклонениям у пациентов с кариотипом XXY (Giedd et al., 2007).
Начиная со среднего детского возраста отмечается тенденция к удлинению ног, а размах рук часто превышает рост. Пенис может быть уменьшенного или нормального размера, яички почти всегда уменьшены, а продукция спермы нарушена. У многих пациентов отмечается увеличение молочных желез, а частота рака груди при данном заболевании выше, чем у здоровых мужчин. Часто пациенты характеризуются как застенчивые, неуверенные в себе. У большинства отмечаются легкие/умеренные проблемы социального взаимодействия и склонность избегать коллективной деятельности. У небольшого количества пациентов отмечается аутизм. Результаты одного проспективного исследования позволяют предположить, что взрослые пациенты с синдромом Клайнфелтера менее сексуально активны и более склонны к подчинению в сексуальной жизни, чем другие мужчины (Schiavi et al., 1988).
Лечение тестостероном следует начинать в препубертатном периоде под наблюдением эндокринолога, являющегося специалистом по синдрому Клайнфелтера. Интрацитоплазматическая инъекция спермы позволяет осуществлять детородную функцию, даже в случае, если в эякуляте сперматозоиды отсутствуют. Даже в случае стойкой азооспермии возможно выделение сперматозоидов из биптатов яичек и реализации, беременности и деторождения (Lanfranco et al., 2004).
Синдром Тернера
Многие пациенты с синдромом Тернера гиперактивны в раннем детском возрасте, но начиная с подросткового возраста склонны к гипоактивности (Swillen et al., 1993). Предполагается взаимосвязь синдрома Х0 с нервной анорексией (в частности, при мозаичном генотипе), но результаты систематического хромосомного исследования не подтверждают данного предположения (Rastam et al., 1991). После подросткового возраста пациенты характеризуются низкорослостью и инфантильным строением наружных и внутренних половых органов. Заместительная терапия эстрогенами необходима, но, тем не менее, не существует четких рекомендаций по поводу возраста начала лечения. Многие женщины с синдромом Тернера выходят замуж и во взрослом возрасте живут нормальной жизнью. Пациентки с синдромом Тернера, получившие Х-хромосому от матери, имеют больше проблем в социальном взаимодействии, чем женщины, получившие Х-хромосому от отца (Skuse et al., 1997).
г) ХХХ синдром. Девочки с кариотипом XXX составляют 0,1% новорожденных девочек. У таких детей имеется повышенный риск нарушений речи и обучения, IQ близок к нижней границе нормы, что приводит к необходимости применения специальных образовательных программ, тем не менее, нарушения чтения и письма соответствуют общему уровню IQ. Пациентки характеризуются застенчивостью, незрелостью и различными поведенческими проблемами (Linden et al., 1988). Характерен высокий рост и плохая координация движений. Данные отклонения в сочетании со странным поведением приводят к высокой частоте обращения в специализированные учреждения.
Синдром дисомии по Y-хромосоме (47,XYY) встречается с частотой 1:1000 новорождённых мальчиков. Большинство мужчин с таким набором хромосом не отличаются от нормальных индивидов по физическому и умственному развитию, имеют рост немного выше среднего. Заметных отклонений ни в половом развитии, ни в гормональном статусе, ни в плодовитости у большинства XYY-индивидов нет. Не исключены некоторые особенности поведения таких лиц: при соответствующих условиях они склонны к агрессивным и даже криминальным поступкам.
Наиболее просматриваемые статьи:
Читайте также: