
Синдром ди джорджи кратко
Обновлено: 02.05.2024
Синдром Ди Джорджи (вело-кардио-фациальный синдром, синдром делеции 22q11) - этиология, клиника, диагностика
Синдром Ди Джорджи (вело-кардио-фациальный синдром, синдром делеции 22q11) связан с дефектом развития четвертого глоточного кармана и четвертой жаберной дуги. Данный синдром имеет генетически неоднородный характер с частичной моносомией проксимального плеча 22-й хромосомы (при микроскопическом исследовании выявляется в трети случаев) и высокой частотой делеций (88%), выявленных по результатам FISH-исследований (Driscoll et al., 1993). В редких случаях заболевание сочеталось делецией 17р и 10p (Levy-Mozziconacci et al., 1994).
Уровень распространения синдрома делеции 22q11 (22Q11DS оценивается приблизительно как 3 случая на 10000 новорожденных (Oskarsdottir 2005) и возможно несколько чаще встречается среди девочек (личные неопубликованные данные).
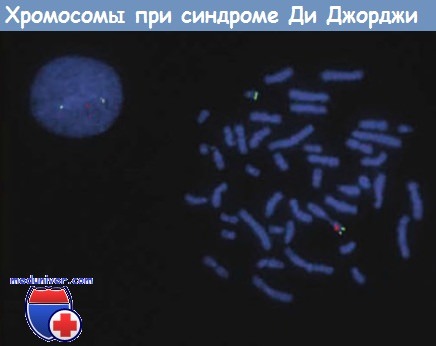
Анализ методом FISH обеих метафазных хромосом и интерфазной клетки пациента с синдромом Ди Джорджи,
демонстрирующий делецию зонда TUPLE1 (официальное название — HIRA), который локализуется на хромосоме 22q11.2.
Красным светится зонд TUPLE1, зеленым — контрольный зонд, локализующийся на 22q. Метафазная пластинка содержит одну 22-ю хромосому с зеленым и красным сигналами.
Другая 22-я хромосома содержит только зеленый сигнал, красный сигнал отсутствует в связи с делецией на этой хромосоме.
На интерфазной клетке также определяются две области гибридизации: зеленая и красная, что иллюстрирует делецию на хромосоме 22q11.2.
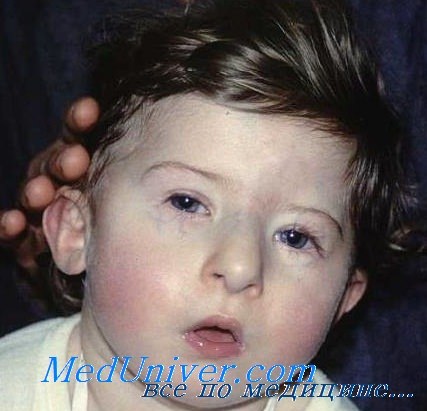
а) Клинические проявления. Тимус и паратиреоидные железы отсутствуют или эктопированы, также часто отмечается мальформация крупных сосудов основания сердца. Отсутствие тимуса может сопровождаться тяжелым иммунодефицитом, а отсутствие паращитовидных желез—тяжелыми гипокальциемическими судорогами в период новорожденности и позже. Гипокальциемия связана с паратиреоидным гормоном. Часто встречается расщелина неба, микрогнатия, низко расположенные уши и гипертелоризм (Conley et al., 1979).
Частично совпадающий фенотип, известный как вело-кардио-фациальный синдром или синдром Шпринтцена, проявляется сходными аномалиями сердца, дисморфизмом лица, расщелиной неба и задержкой умственного развития, иногда сопровождается поздним развитием психиатрических отклонений и также сочетается с делецией 22q11. Также может отмечаться гипоплазия мозжечка (Devriendt et al., 1996).
Синдром делеции сегмента 22q11 проявляется достаточно типичным поведенческим фенотипом. Отмечается задержка развития разговорной речи, но некоторые вербальные навыки, в частности словарный запас, имеют тенденцию к лучшему развитию, чем функции, связанные с достижением школьного возраста. Коэффициент IQ обычно составляет около 70, у отдельных пациентов превышает 100 и (очень редко) не достигает 40. Неуклюжесть движений является типичным проявлением данного заболевания, и клинические проявления синдрома делеции сегмента 22q11 часто полностью соответствуют критериям нарушения развития координации.
Синдром Ди Джорджи
Даже в случаях, когда признаки аутизма отсутствуют, у пациентов отмечаются социальные проблемы и проблемы общения. Данные отклонения часто сопровождаются растягиванием слов, проблемами артикуляции и бедной мимикой, характерной для синдрома делеции сегмента 22q11, так как во многих случаях отмечается небно-глоточная недостаточность. Депрессивное настроение часто отмечается, начиная с подросткового возраста. Психозы могут развиваться в позднем подростковом возрасте и раннем взрослом возрасте, даже в случаях с минимальными психиатрическими отклонениями в детском возрасте (Sobin et al., 2005). СДГВ и расстройства группы аутизма при синдроме делеции сегмента 22q11 могут быть результатом патологии головного мозга с уменьшением серого вещества коры, большого хвостатого ядра и аномалии белого вещества лобных долей и мозжечка. В подростковом и взрослом возрасте увеличивается риск развития шизофрении (Niklasson et al., 2002).
б) Лечение синдрома Ди Джорджи. Как и в случае других синдромов с поведенческим фенотипом важен ранний диагноз и обучение семьи психологической помощи. СДГВ в некоторых случаях успешно поддается лечению стимуляторами и методами когнитивно-поведенческой терапии в сочетании со специальными программами образования. Ингибиторы захвата серотонина или комбинированного захвата серотонина и норадреналина можно попробовать при длительных периодах депрессивного настроения или дистимии. Психоз (в случае его развития) может с трудом подаваться лечению, а типичные и атипичные нейролептики, несмотря на их эффективность в отношении психотических симптомов, а иногда и слуховые галлюцинации, часто менее эффективны, чем в случае других психозов у молодых людей.
Синдром Ди Джорджи еще в некоторых источниках указывают как синдром Ди Георга. Впервые патология была описана в 1965 году американским педиатром и эндокринологом — Анджело Мария Ди Джорджи (фото). По международной классификации десятого пересмотра (МКБ-10) данному иммунодефициту был присвоен код D82.1 и указан как синдром дивертикула глотки, вилочковой железы: алимфоплазия, аплазия либо гипоплазия с иммунной недостаточностью.

Синдром Ди Джорджа как первичный иммунодефицит является редким врожденным заболеванием (1 случай на 4-6 тыс. человек) и относится к идиопатическому изолированному гипопаратиреозу – состоянию, для которого характерно снижение выработки паратгормонов и гипокальциемия. Для синдрома свойственно множество фенотипов поражения, осложнений и присоединения сопутствующих нарушений.
Патогенез
Для синдрома Ди Георга характерна аплазия или гипоплазия вилочковой железы (тимуса), а также агенезия или дисгенезия паращитовидных желез в результате нарушений закладки и эмбриональной дифференцировки 3-4 жаберных (глоточных) карманов. Это вызывает резкое снижение популяции Т-лимфоцитов, нарушение их дифференцировки и как следствие — иммунологическую недостаточность. У детей могут развиваться различные врождённые аномалии крупных сосудов, например, дефекты аорты, межжелудочковой перегородки или синий порок сердца.
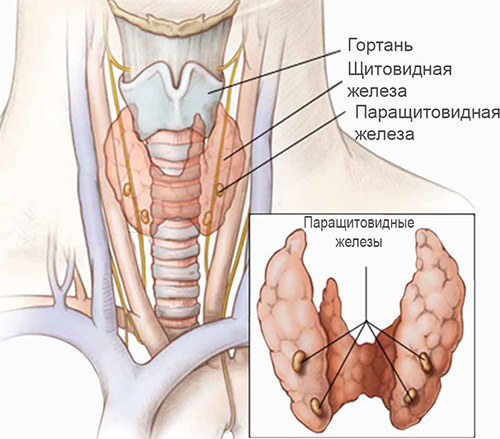
Расположение паращитовидных и щитовидных желез
Классификация
В зависимости от спектра клинических проявлений и врожденных аномалий синдром Ди Георга может быть полным и частичным, к примеру, протекать как изолированная недостаточность паращитовидных желёз либо врождённое отсутствие паращитовидных (околощитовидных) желез, которые приводят к гипокальциемическим судорогам, наблюдающимся у новорожденных в виде неонатальной тетании. Нарушения закладки тимуса приводят к развитию различных инфекционных заболеваний и обычно вызваны иммунологической недостаточностью.
Достаточно часто болезнь протекает в менее тяжелой форме и является наиболее распространенной причиной умственной отсталости с малым количеством других проявлений, поэтому может быть формально не диагностирована.
Причины
У синдрома Ди Джорджи или врождённой аплазии тимуса и паращитовидной железы генетическая причина развития. Патология развивается в результате делеции центрального участка более длинного плеча двадцать второй хромосомы, поэтому еще обозначается как синдром 22q 11.2. Однако, в некоторых случаях аналогичная клиническая картина наблюдалась и после других хромосомных перестроек – транслокаций и микроделеций, развивающихся в таких хромосомах как ТВХ1, 10р13, 17р13, 18q21 и пр. Эти мутации спорадические в 90% и обычно происходят на этапе мейоза спермато— или овогенеза.
Для данного синдрома дисэмбриогенеза 3-4 жаберной дуги — фенотипа CATCH 22 характерно наследование по аутосомно-доминантному типу, которое наблюдается лишь в 5-10% случаев, но при этом не исключается возможность аутосомно-рецессивного типа наследования с разной экспрессивностью.
Симптомы врождённой аплазии тимуса и паращитовидных желёз (Синдром Ди Джорджи)
Синдром Ди Джорджи – это первичный иммунодефицит, вызванный врожденным отсутствием или наличием аномалий развития тимуса и паращитовидных желез, который отличается триадой клинических симптомов, включающей:
- различные врожденные пороки сердца, дефекты крупных сосудов, носа, рта, ушей, что в результате дает специфические черты лица (как на фото): гипертелоризм (увеличенное расстояние между глазами), микрогнатия (недоразвитие нижней челюсти), низкое расположение ушных раковин;
- первичный иммунодефицит – нарушен как клеточный, так и гуморальный ответ иммунитета в результате аплазии вилочковой железы и невозможности обеспечения нормального развития Т-клеток;
- прогрессирующая гипокальциемия (пониженная концентрация кальция в кровотоке) – вызвана гипопаратиреозом, сопровождается судорожным синдромом и развитием скелетных аномалий.
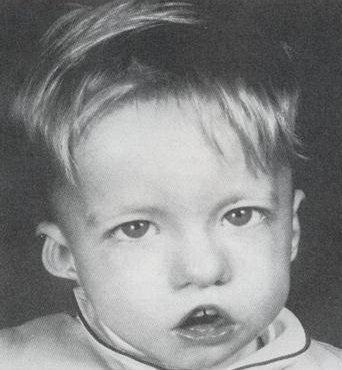
Ребенок со специфическими чертами, характерными для синдрома Ди Джорджи
Особенности синдрома могут широко варьироваться даже в пределах одной семьи и затрагивать различные системы органов. Сопутствующими проблемами становится:
- нарушение работы почек и их атрофия, развитие нефрокальциноза, гидронефроза и пр.;
- проблемы с пищеварением и перистальтикой ЖКТ;
- дефицит гормона роста;
- проблемы с речью;
- психические расстройства, в том числе шизофрения;
- потеря слуха (проводящая и нейросенсорная обычно вызвана черепно-лицевыми синдромами);
- нарушения координации, которые могут быть вызваны гипоплазией мозжечка;
- синюшность кожных покровов в результате плохого кровообращения; ;
- частые переломы костей;
- болезнь Грейвса; .
Анализы и диагностика
Для подтверждения диагноза синдром Ди Джорджа необходимо выявление агенезии или дисгенеза паращитовидных желёз, аплазии вилочковой железы, иммунологической недостаточности, черепно-лицевых дисморфий (микрогнатии, гипертелоризма, антимонголоидного разреза глаз, расщелины губы и нёба, деформированных и/или низко расположенных ушных раковин) и прочих типичных аномалий развития. При этом оказываются эффективными различные исследования, включая УЗИ, МРТ и пр., а также используются методы генетического тестирования, к примеру, FISH (метод флуоресцентной гибридизации in situ).
Наиболее яркими проявлениями является гипопаратериоз и молочница. Также при плановом обследовании выявляются дефекты аорты, тетрада Фалло, катаракта, паховая грыжа и тому подобное. При помощи серологических исследований можно выявить лимфоцитопению, гипокальциемию, гипоγ-глобулинемии. Благодаря иммунологическим методам удается обнаружить нарушения трансформации лимфоцитов и их функциональной активности, в 20% случаев наблюдается сниженное количество Т-клеток. После иммунологических исследований важно провести дифференциальный диагноз с прочими первичными иммунодефицитами, таким как: синдром Вискотта-Олдрича, Брутона.
Лечение
Так как недуг неизлечим больным обычно назначают симптоматическое и комплексное поддерживающее лечение. Основными приемами считается соблюдение диеты, стерильные условия, прием кальция, комплексов витамин, антибиотиков, противогрибковых, иммуномодулирующих, седативных препаратов и пр.

Синдром Ди Джорджи – патология внутриутробного развития, которая сопровождается нарушением в 22 хромосоме. На стадии формирования третьей и четвертой жаберных дуг происходит их недоразвитие и как следствие, в дальнейшем прогрессирует патологическое состояние тимуса и паращитовидных желез
Одной из причин этого заболевания врачи называют перенесенные во время беременности инфекции, а также сахарный диабет у матери. Около 15% причин приходится на наследственные факторы. Впервые синдром Ди Джорджи был описан американским врачом-педиатром Анджело Ди Джорджи в 1965 году. Автор определил его как врожденное недоразвитие вилочковой и паращитовидных желез.
Основная причина высокой детской смертности с данным заболеванием заключается в патологических состояниях сердечно-сосудистой, иммунной и эндокринной системы. Сразу же после рождения ребенка его организм подвержен вирусной, бактериальной и грибковой инфекции, которая на форме врожденных пороков сердца и сбоях в работе вилочковой железы существенно увеличивает риск летального исхода.
К сожалению, не существует специальной этиологической терапии, которая позволяет повлиять на генетическую мутацию синдрома Ди Джорджи. Все лечение направлено на преодоление симптомов и последствий заболевания. В качестве поддерживающего метода для нормализации уровня фосфора и кальция применяется заместительное лечение.
Адекватных методов профилактики иммунодефицитного состояния не разработано, кроме отдельных случаев пересадки тимуса или костного мозга. Поэтому прогноз обычно крайне неблагоприятный и без своевременной медикаментозной поддержки большинство детей не доживают больше первого года жизни. Даже при достаточной терапии, качество жизни людей с этой патологией крайне низкое и зависит от грамотной стратегии врачей.
Синдром Ди Джорджи связан с гипо- или аплазией тимуса и паращитовидных желез, приводящей к Т-клеточному иммунодефициту и гипопаратиреозу. У младенцев с синдрома Ди Джорджи наблюдается низкое расположение ушных раковин, срединная расщелина лица, слабое недоразвитие нижней челюсти, гипертелоризм, короткий губной желобок, задержка развития и врожденные заболевания сердца. Диагноз ставят на основании клинических данных и включает оценку иммунной функции и функции паращитовидных желез, а также хромосомный анализ. Лечение включает поддерживающую терапию и, в тяжелых случаях, трансплантацию тимуса или стволовых клеток.
Синдром Ди Джорджи может быть
Частичным: присутствуют некоторые функции Т-клеток
Полным: отсутствие функции Т-клеток
Симптомы и признаки синдрома Ди Джорджи

У младенцев с синдрома Ди Джорджи наблюдается низкое расположение ушных раковин, срединная расщелина лица, слабое недоразвитие нижней челюсти, гипертелоризм, короткий губной желобок, задержка развития и врожденные заболевания сердца Краткий обзор врожденных сердечно-сосудистых аномалий (Overview of Congenital Cardiovascular Anomalies) Врожденный порок сердца является наиболее распространенной врожденной аномалией, которая возникает у почти 1% живорожденных (1). Среди врожденных дефектов врожденный порок сердца является ведущей. Прочитайте дополнительные сведения (например, коарктация дуги аорты, общий артериальный ствол, тетрада Фалло, дефекты межпредсердной или межжелудочковой перегородки). У детей имеет место гипо- или аплазия тимуса и паращитовидных желез, ведущие к Т-клеточной недостаточности и гипопаратиреозу Гипопаратиреоз Гипокальциемию диагностируют при концентрации общего кальция в сыворотке 8,8 мг/дл ( 2,20 ммоль/л) на фоне нормального уровня белков в плазме или при концентрации ионизированного кальция в сыворотке. Прочитайте дополнительные сведения .
Рецидивирующие инфекции начинаются сразу после рождения, но степень иммунодефицита значительно варьирует, функция Т-лимфоцитов может спонтанно улучшаться. Гипокальциемические судороги наблюдаются в течение 24–48 часов после рождения.
Прогноз зачастую зависит от тяжести пороков сердца.
Диагностика синдрома Ди Джорджи
Оценка иммунной функции по уровням иммуноглобулина (Ig), количественное определение титров вакцин и субпопуляций лимфоцитов
Диагностика синдрома Ди Георге основана на клинических данных.
Проведение подсчета абсолютного содержания лимфоцитов с последующим подсчетом В- и Т-клеток и оценком подпопуляций лимфоцитов в случае, если выявлена лейкопения; проводят анализ крови для подсчета Т-клеток и функции паращитовидной железы. Измерение уровней Ig и титров вакцин. При подозрении на полный синдром Ди Джорджи следует провести исследование Т-рецепторных эксцизионных колец (TREC).
Рентгенография органов грудной клетки в боковой проекции может помочь обследовать тени от тимуса.
Флуоресцентная гибридизация in situ (FISH) выявляет делецию локуса 22q11 в 22-й хромосоме; может быть проведено стандартное хромосомное тестирование для выявления других нарушений.
При подозрении на полный синдром Ди Джорджи следует провести эхокардиографию. Если у пациентов присутствует цианоз, может быть необходимо проведение катетеризации сердца.
Поскольку в большинстве случаев заболевание является спорадическим, скрининг родственников не является необходимым.
Лечение синдрома Ди Джорджи
Частичный синдром: добавки кальция и витамина D
Полный синдром: трансплантация культивированной ткани вилочковой железы или гемопоэтических стволовых клеток
При частичном синдроме Ди Джорджи гипопаратиреоз лечится препаратами, содержащими кальций, и витамином D; продолжительность жизни не нарушается.
Справочные материалы по лечению
1. Davies EG, Cheung M, Gilmour K, et al: Thymus transplantation for complete DiGeorge syndrome: European experience. J Allergy Clin Immunol140: 1660–1670.e16, 2017.
Читайте также: