
Механизм активации и инактивации g белков кратко
Обновлено: 05.07.2024
Сигнальные G-белки являются универсальными посредниками при передаче гормональных сигналов от рецепторов клеточной мембраны к эффекторным белкам , вызывающим конечный клеточный ответ.
Когда семидоменная рецепторная молекула , локализованная в мембране сенсорной клетки, активируется какими-то изменениями во внешней среде, она претерпевает конформационные изменения. Последние детектируются G-белками , связанными с мембраной, которые, в свою очередь, активируют эффекторные молекулы в мембране. Часто это приводит к выделению вторичных мессенджеров в цитозоль. G-белки, участвующие в передаче сигнала, являются членами большого надсемейства гуанин-связывающих белков. G- белки - это прецизионные регуляторы, включающие или выключающие активность других молекул.
Примерно 80% первичных мессенджеров (гормоны, нейротрансмиттеры, нейромодуляторы) взаимодействуют со специфическими рецепторами, которые связаны с эффекторами через G-белки.
G-белки -белки, связывающие гуанозиновые нуклеотиды. G-белки, ассоциированные с рецепторами, связаны с мембраной . G-белки биологических мембран имеют гетеротримерную структуру. Они состоят из большой альфа-субъединицы (около 45 килодальтон - кДа), а также меньших бета- и гамма-субъединиц. Альфа-субъединица обладает ГТФ-азной активностью, в неактивной (выключенной) форме она связывает молекулу ГДФ на активном сайте. Субъединицы бета и гамма связаны между собой, и в физиологических условиях не могут быть диссоциированы. В неактивном состоянии бета-гамма-комплекс непрочно связан с альфа-субъединицей. Гамма-субъединица связана с цитоплазматическим листком биологической мембраны геранил-гераниловой цепью (20 атомов углерода в цепи) , близкой по структуре к холестерину. Альфа-субъединица также связана с мембраной жирной кислотой с длиной цепи в 14 атомов углерода ( миристоевая кислота ). Такие связи обеспечивают то, что комплекс G-белка удерживается в плоскости мембраны, но в то же время способен легко двигаться в этой плоскости. Малые GTPaзы участвуют в контроле фундаментальных свойств клетки - полярности формы и процессов деления и дифференцировки. G-белки обычно регулируют более специализированные сигналы - продукцию вторичных мессенджеров. И те и другие способны гидролизовать GTP и таким образом выключать сигнал.
Поскольку бета- и гамма- субъединицы G-белков чрезвычайно консервативны, G-белки принято различать по их альфа-субъединицам. G-белки, стимулирующие аденилатциклазу (Gs) или участвующие в фототрансдукции (Gt, трансдуцин) служат субстратами для АДФ- рибозилирования, катализируемого холерным токсином по одному из остатков аргинина, что приводит к блокированию деактивации этих белков. Gs, G-белок, ингибирующий аденилатциклазу (Gi) и G-белок с пока еще неизвестной функцией (Gо) АДФ-рибозилируются коклюшным токсином по остатку цистеина, расположенному у C-конца. Эта модификация препятствует взаимодействию между G-белком и рецепторами. Определена последовательность G-белка крысы (Gx), который оказался нечувствительным к коклюшному токсину.
G-белки это - регуляторные белки, связывающие при активации ГТФ . Лучше всего изучены G-белки, стимулирующие и ингибирующие аденилатциклазу (Gs-белки и Gi-белки соответственно). Бета1-адренорецепторы , бета2-адренорецепторы и D1-рецепторы сопряжены с белком Gs, и поэтому стимуляция этих рецепторов сопровождается активацией аденилатциклазы и повышением внутриклеточной концентрации цАМФ - классического второго (внутриклеточного) посредника. Конечный ответ в разных клетках различен и зависит от того, что представляет собой эффекторные фермент .Связывание агониста (гормона, нейромедиатора и др.) с соответствующим рецептором приводит к белок-белковому взаимодействию между рецептором и G-белком и ускоряет диссоциацию ГДФ. В результате образуется короткоживущий комплекс агонист - рецептор - G-белок, не связанный ни с каким нуклеотидом. Связывание с этим комплексом молекулы ГТФ снижает сродство рецептора к G-белку, что приводит к диссоциации комплекса и высвобождению рецептора. Потенциально рецептор может активировать большое количество молекул G-белка, обеспечивая, таким образом, высокий коэффициент усиления внеклеточного сигнала на данном этапе. Активированная альфа-субъединица G-белка (альфа* ГТФ Мg). [ Bourne, ea 1997 ] диссоциирует от бета-гамма-субъединиц и вступает во взаимодействие с соответствующим эффектором, оказывая на него активирующее или ингибирующее воздействие.
Альфа-субъдиница с присоединенным с ней ГТФ способна взаимодействовать с эффектором в мембране - ферментами, такими, как аденилатциклаза , или, возможно, ионными каналами . Фермент может активироваться или ингибироваться, а ионный канал - открываться или закрываться. Взаимодействие с эффектором, однако, длится до тех пор, пока альфа- субъединица, являющаяся ГТФ-азой, удерживает ГТФ. Так что, очень вскоре присоединенный ГТФ гидролизуется до ГДФ. Когда это происходит, альфа- субъединица снова меняет свою коонформацию и теряет способность активировать эффектор.
У любых клеток есть мембранные белки-рецепторы. Это трансмембранные белки, пересекающие липидный бислой один или несколько раз. В состав многих из них входят олигосахариды (такие рецепторы правильнее называть гликопротеидами).
Такие белки присутствуют не только на наружной мембране, но и на многих внутриклеточных мембранах. Например, рианодиновые рецепторы и рецепторы инозитолтрифосфата есть на мембране эндоплазматического ретикулума.
Мембранные рецепторы связывают сигнальное вещество (лиганд) и при этом изменяют свою конформацию. Часть из них одновременно являются ионными каналами; при связывании лиганда канал может открываться или закрываться. Такие белки называются ионотропные рецепторы. Другие рецепторы при связывании лиганда запускают какую-нибудь химическую реакцию на внутренней стороне мембраны (так что они являются одновременно ферментами или регуляторными белками); такие белки называются метаботропные рецепторы.
Рецепторы, сопряженные с G-белками - это семиспиральные (семь раз пересекающие мембрану в виде альфа-спиралей) белки. Рассмотрим механизм их действия на примере бета-2 адренорецепторов. Это - один из типов адренорецепторов, чувствительный в основном к андреналину (норадреналин действует на них в меньшей степени).

Выявленная с помощью рентгеноструктурного анализа структура β2-адренорецептора, связанного с одним из искусственных лигандов
При действии адреналина на эти рецепторы гладкие мышцы бронхов и кровеносных сосудов скелетных мышц расслабляются, а в клетках печени усиливается распад гликогена (гликогенолиз), и образующаяся глюкоза выходит в кровь. С данным типом рецепторов связан Gs-белок. Этот белок, как и другие G-белки, состоит из трех субъединиц (полипептидных цепей) - α,β, и γ. Он "приделан" к внутренней стороне мембраны с помощью двух хвостов жирных кислот и свободно передвигается в плоскости мембраны. С α-субъединицей неактивного Gs-белка связана молекула ГДФ. Когда на бета-2 рецептор действует адреналин, рецептор активируется (меняет свою конформацию) и активирует Gs-белок. В результате α-субъединица отделяется от βγ-субъединицы и обменивает молекулу ГДФ на молекулу ГТФ. Такая активная α-субъединица соединяется с трансмембранным белком-ферментом аденилатциклазой, активируя его.
Этот фермент осуществляет синтез циклического аденозинмонофосфата (цАМФ) из АТФ. цАМФ - один из универсальных вторичных посредников, используемых для передачи сигнала в клетках. В данном случае цАМФ активирует одну из протеинкиназ - протеинкиназу А (РКА). Этот фермент состоит из четырех субъединиц - двух регуляторных и двух каталитических. При связывании четырех молекул цАМФ регуляторные субъединицы отделяются от каталитических, которые при этом активируются. В клетках печени РКА фосфорилирует другую протеинкиназу - киназу фосфорилазы. Киназа фосфорилазы, в свою очередь, фосфорилирует фосфорилазу гликогена. Под действием фосфорилазы происходит фосфоролиз гликогена [1]. В результате образуется глюкоза,которая через белки-переносчики выходит из клеток печени в кровь и потребляется активно работающими при стрессе органами - в первую очередь скелетными мышцами.
Зачем же нужна такая сложная и многоступенчатая система передачи сигнала? Во-первых, на первых этапах сигнал передается с внешней стороны мембраны (на которую действует гормон) на внутреннюю, где происходит синтез цАМФ. Хорошо растворимая, гидрофильная молекула цАМФ быстро диффундирует по всей клетке и передает сигнал во все ее участки. Во-вторых, на каждом этапе сигнализацию можно регулировать. Но главный смысл многоступенчатой передачи - в том, что на большинстве этапов происходит усиление сигнала. Так, за время активности рецептора он может активировать множество молекул G-белка. Каждый Gs-белок активирует одну молекулу аденилатциклазы, но аденилатциклаза синтезирует тысячи молекул цАМФ. 4 молекулы цАМФ активируют всего две каталитических субъединицы РКА, но те могут фосфорилировать множество молекул киназы фосфорилазы, и т.п. В результате такой системы многократного усиления под действием одной молекулы адреналина в клетке печени образуется около 10.000.000 молекул глюкозы.
Когда стресс прошел, уровень секреции адреналина снижается. Из крови адреналин быстро выводится через почки и перестает действовать на рецепторы. После этого инактивируется Gs-белок. G-белки обладают ГТФ-азной активностью: α-субъединица расщепляет связанную с ней молекулу ГТФ до ГДФ (и фосфата), после чего связывается с βγ-субъединицей и переходит в неактивное состояние. таким образом, G-белок действует как автоматический "молекулярный выключатель". Уровень цАМФ в клетке понимается до исходного за счет того, что цАМФ расщепляет особый фермент - фосфодиэстераза. В результате каталитические субъединицы РКА объединяются с регуляторными и инактивируются. Инактивацию киназы фосфорилазы и фосфорилазы гликогена осуществляют ферменты протеинфосфатазы, отщепляющие от этих ферментов фосфатные группы. Так распад гликогена прекращается.
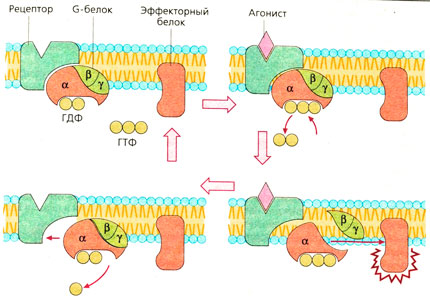
Механизм передачи сигнала одинаков у всех G-белок-связанных рецепторов (А). После присоединения агониста к рецептору изменяется конформация рецепторного белка. а-Субъединица отдает ГДФ и присоединяет ГТФ, затем отделяется от двух других субъединиц, вступает в контакт с эффекторным белком и изменяет его функциональное состояние. И β, и у-субъединицы способны связываться с эффекторными белками. а-Субъединица обеспечивает медленный гидролиз связанного ГТФ до ГДФ. Ga-ГДФ не имеет сродства к эффекторным белкам и вновь воссоединяется с β, у-субъединицами (А).
G-Белки могут диффундировать (латерально) в мембрану; они не связаны с определенным типом рецепторов. Тем не менее между типами рецепторов и типами G-белков имеется взаимосвязь (Б). а-Субъединицы G-белков различаются по сродству и типу воздействия на эффекторные белки. Ga-ГТФ Gg-белка стимулирует аденилатцик-лазу, в то время как Ga-ГТФ Gj-белка ее ингибирует. К G-белок-связанным рецепторам относятся мускариновые холинорецепторы, рецепторы к норадреналину, адреналину, допамину, гистамину, морфину, простагландинам, лейкотриенам и другим веществам и гормонам.
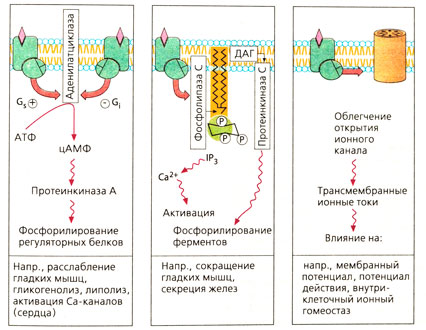
Эффекторными белками для G-белка являются аденилатциклаза (АТФ - внутриклеточный переносчик цАМФ), фосфолипаза С (фосфатидилинозитол -> внутриклеточные переносчики инозитолтрифосфат и диацилглицерин) и некоторые белки ионных каналов (Б).
От концентрации цАМФ в клетке зависит множество функций, так как цАМФ повышает активность протеинкиназы А, которая фосфорилирует регуляторные белки. Кроме того, повышение концентрации цАМФ приводит к расслаблению гладкой мускулатуры, повышению силы сердечных сокращений, усилению гликогенолиза и липолиза. Фосфорилирование белков Са-каналов способствует их открытию при деполяризации мембраны. Необходимо отметить, что цАМФ инактивируется фосфодиэстеразой. Поэтому ингибиторы этого фермента поддерживают высокую клеточную концентрацию цАМФ и оказывают сходное с адреналином действие.
Кроме того, рецепторный белок может фосфорилироваться и вследствие этого теряет способность активировать G-белок. Данный механизм лежит в основе снижения чувствительности клетки в результате длительной стимуляции рецептора под действием агониста.
Активация фосфолипазы С приводит к расщеплению фосфолипидов мембран (фосфатидилинозитол-4,5-бифосфата) с образованием инозитолтрифосфата (1Р3) и диацил глицерина (ДАГ). Инозитол стимулирует выход Са2+ из депо, что ведет к сокращению гладкой мускулатуры, расщеплению гликогена или экзоцитозу. Диацилглицерин стимулирует протеинкиназу С, которая фосфорилирует определенные серин- и треонинсодержащие ферменты.
Некоторые G-белки воздействуют на белки каналов и способствуют открытию каналов. Таким образом активируются К+-каналы (действие ацетилхолина на синаптическом уровне; влияние опиоидов на передачу возбуждения в нервных клетках).

В таблице приведены примеры подобных рецепторных систем. Очевидно, что большое количество первичных мессенджеров регулируют лишь несколько вторичных. Таким образом, следует понимать, что специфичность действия первично связывающихся лигандов определяется локализацией рецептора на определенных клетках, в которых вторичные мессенджеры вызывают экспрессию белков, специфичных для данной клетки.
Открыто более 1000 генов, кодирующих рецепторы, связанные с G-белками. Эти рецепторы являются мономерными гликопротеинами с относительно сходным аминокислотным составом. Для них характерны 7 доменов, имеющих в составе полипептидной цепи гидрофобные аминокислоты. Эти домены образуются петлями внутри мембраны с одним концом, выступающим наружу, а другим -внутрь цитоплазмы.

Эндогенные лиганды связываются на внешнем участке рецептора. Небольшие по размеру, например амины, присоединяются к участкам внутри различных трансмембранных областей, тогда как крупные, например полипептиды, менее способные проникать в гидрофобные регионы, связываются на экстраклеточной петле N-концевого участка рецептора.
G-Белки присоединяются к внутриклеточному сегменту рецептора внутри 3-й петли между 5-м и 6-м регионами. Хотя механизм эффекта, возникающего при соединении с агонистом, не до конца ясен, предполагается, что этот акт стабилизирует рецептор в конформации, позволяющей ему взаимодействовать с тримером G-белков, активируя их и последующие эффекторные события.
Эффект агониста обычно имеет лишь ограниченную продолжительность, что определяется несколькими процессами. Большинство связанных с G-белкам и рецепторов имеет остатки серина и треонина в цитоплазматической петле и в С-концевом участке, которые могут быть фосфорилированы несколькими киназами, что ограничивает взаимодействие между рецептором и G-белком. Данный процесс называется десенситезацией рецептора и позволяет клетке ответить на весьма широкие диапазоны концентраций, стимулирующих экстраклеточных агентов. После продолжительного воздействия агониста число рецепторов на плазматической мембране может также регулироваться процессом интернализации (осуществляемым в некоторых случаях катаболизмом), или down-регуляцией рецептора. Хотя вызывающие ее сигналы не ясны, представляется, что они отличны от контролирующих десенситезацию.
Осуществляют трансдукцию сигнала от мембранных рецепторов к эффекторным ферментам и ионным каналам. Каждый из этих белков состоит из трех отдельных субъединиц, обозначаемых α, β, у в порядке снижения молекулярной массы; α-субъединица тримера связывает гуаниннуклеотиды и является главным посредником влияния G-белков на их эффектор. Основной функцией β- и γ-субъединиц тримера является поддержка взаимодействия α-субъединицы с плазматической мембраной и рецепторами, однако они способны и к прямой регуляции эффектора.

G-белки, участвующие в трансмембранном переносе сигнала, регулируются общими механизмами. На рисунке представлен их цикл активации и инактивации. В базальном состоянии три субъединицы G-белков связаны вместе и с гуанозиндифосфатом (ГДФ), присоединенным к α-субъединице. Когда агонист соединяется с рецептором, ГДФ отделяется, и освободившееся на α-субъединице место занимается гуанозинтрифосфатом (ГТФ), в избытке присутствующем в цитоплазме.
Появление этой связи стимулирует G-белки, в результате α-субъединица отделяется от рецептора и от β, γ-субъединиц и связывается с эффектором. Спустя несколько секунд имеющаяся в α-субъединице ГТФаза гидролизует связанную ГТФ до ГДФ, что приводит к инактивации субъединицы. Связанная с ГДФ α-субъединица отделяется от эффектора и вновь ассоциируется с β-, γ-комплексом G-белков, становясь способной к новому циклу активации рецептором.
Функциональные различия между членами семейства G-белков первично определяются отличиями в α-субъединицах. Номенклатура G-белков изначально строилась в соответствии с их функцией. Так, обозначения Gs и Gi приняты для стимулирующих и ингибируюших аденилатциклазу G-белков соответственно. Обозначение Gi было введено раньше, когда впервые открытый в сетчатке G-белок назывался трансдуцином.
Несовершенство этой номенклатуры стало очевидным после выделения и клонирования G-белков с невыясненной функцией. Так появились обозначения Go, Gq , затем G11, G12 и др. Некоторые G-белки экспрессируются лишь в определенных типах клеток и высокоспециализированы, например Gt обнаружен только в палочках и колбочках сетчатки, где активирует цГМФ-специфичную фосфодиэстеразу. Другие G-белки, наоборот, экспрессируются в большинстве тканей и имеют множественную функцию, например, Gi присутствует в большинстве клеток, ингибируя аденилатциклазу, но способен и к прямому воздействию на некоторые ионные каналы.
Влияние большинства G-белков на эффекторы — стимулирующее, однако некоторые их ингибируют. Например, G0 ингибирует Са2+-каналы в мозге и сердце; Gs стимулирует, a Gi ингибирует аденилатциклазу. Последние два G-белка в свою очередь регулируются различными группами рецепторов, которые часто представлены в одной клетке. Результат одновременной активации стимулирующих и ингибирующих рецепторов — уменьшение и смягчение аденилатциклазного ответа.
Эта двойная регуляция в дополнение к взаимодействию других следующих за белками компонентов сигнальной системы обеспечивает интегрированный ответ клетки на многочисленные стимулы.
Эффекторные ферменты, регулируемые G-белками. Из трех компонентов, обеспечивающих сопряженное с G-белками проведение сигнала, эффекторные звенья наиболее сложно изучать на молекулярном уровне. Только недавно некоторые из ферментов этого звена были изолированы и клонированы.
Эффекты гормональных и нсйромелиаторных рецепторов чаще всего реализуются через аденилатциклазу и фосфолипазу С. Другие ферменты, такие, как фосфолипаза А2, продуцирующая арахидоновую кислоту, также, по-видимому, регулируются G-белками, однако эти реакции до конца не ясны.
Циклический АМФ (цАМФ) синтезируется из АТФ встроенными в плазматическую мембрану аденилилциклазными ферментами.
Представляется, что эти ферменты являются большими полипептидами, содержащими два кластера из шести трансмембранных сегментов, разделяющих два сходных каталитических домена. Имеется, по крайней мерс, восемь форм аденилатциклазы. Все они стимулируются Gsα, но отличаются по чувствительности к ингибирующему влиянию Gα-стимуляции кальцийзависимым кальмодулином (calcium/calmodulin) и по эффекту β, у-субъединиц G-белков. Эти дополнительные регуляторы дают возможность интеграции многих сигналов, влияющих на различные системы вторичных мессснджеров внутри одной клетки.

Циклический АМФ проявляет свой эффект в клетке, в основном активируя цАМФ-зависимые протеинкиназы (протеинкиназа А, РКА). Эти тетрамерные ферменты состоят из двух регуляторных и двух каталитических субъединиц. Ферменты активируются, когда две молекулы цАМФ присоединяются к каждой регуляторной субъединице, освобождая каталитические субъединицы из тетрамера. Освободившиеся субъединицы катализируют перенос фосфатной группы из АТФ на специфические сериновые или треаниновые остатки белков-мишеней. Среди них могут быть ферменты, которые участвуют в метаболических процессах клетки, и белки, регулирующие транскрипцию гена. Хорошо изучен, например, активируемый цАМФ метаболический путь, составляющий каскад ферментативных активаций, ведущий к распаду гликогена в печени. Активированная протеинкиназа А фосфорилирует фосфорилазную киназу (phosphorylase kinase), которая в свою очередь фосфорилирует гликогенфосфорилазу — фермент, катализирующий распад гликогена.
Действие цАМФ на транскрипцию генов опосредуется катализируемым протеинкиназой А фосфорилированием белка, известного под названием cAMP response clement-binding protein (CREB), который присоединяется к специфическим коротким последовательностям ДНК, известным как cAMP response elements (CRE). CREB присоединяется к CRE, будучи фосфорилированным протеинкиназой А, что стимулирует транскрипцию генов, содержащих CRE в регуляторных зонах.
Представители семейства G-белков сопрягают различные рецепторы с группой ферментов, известных как фосфолипазы с-р. Эти ферменты относятся к большому семейству фосфолипаз, субстратом для которых являются инозитолфосфолипиды. Сигнальная трансдукция через этот путь влечет последовательность молекулярных событий, сходных с наблюдаемыми при активации адеиилатциклазы. Связывание агониста с рецептором активирует G-белок, который в свою очередь присоединяется к фосфолипазе на внутренней поверхности плазматической мембраны. Активированная липаза быстро превращает фосфатидилинозитолбифосфат (PIP2) в инозитолтрифосфат (IP3) и диацилглицерол. Обе эти молекулы действуют как вторичные мессен-джеры двумя различными путями: IP3 — небольшая водорастворимая молекула, способная быстро диффундировать в цитоплазму и присоединяться к 1РЗ-зависимым кальциевым каналам в гладком эндоплазматическом ретикулуме, освобождая запасы кальция в цитозоль.
Увеличение концентрации Са2+ в цитоплазме инициирует волну Са2+-зависимых реакций в клетке, многие из них опосредуются специфическими Са2+-связывающими белками, из которых наиболее распространен кальмодулин. Са2+-кальмодулин регулирует ряд ферментов, включая Са2+-зависимую АТФазу плазматической мембраны, которая выкачивает кальций из клетки, и, как было сказано раньше, некоторые типы адеиилатциклазы. Большинство эффектов кальция в клетке является результатом активации группы протеинкиназ, известных как Са2+-кальмодулинзависимые протеинкиназы. Эти киназы фосфорилируют сери-новые и треониновые остатки различных белков. Таким образом, вновь физиологический ответ на активацию фосфолипидного вторичного мессенджера в каждой клетке зависит именно от экспрес-сирующихся в ней белков, являющихся мишенью Са2+-кальмодулинкиназ.
Другим молекулярным продуктом гидролиза PIP2 фосфолипазой С является диацилглицерол. Эта липидная молекула остается в плазматической мембране, где совместно с фосфатидилсерином активирует некоторых членов другого семейства серин-треониновых кипаз, известных как протеинкиназа С. Эти растворимые киназы переметаются в мембрану в ответ на увеличение кальция в пи го юле (вызванное освобождением IP3) и затем активируются комбинированным воздействием Са2+ диацилглицерола и фосфатидилсерина. Будучи активированными, эти киназы фосфорилируют специфические для клетки группы субстратных белков, которые включают многие ионные каналы, рецепторы и другие киназы, что в результате увеличивает генную транскрипцию.
Другие процессы сигнальной транедукции, регулируемые G-белками. В дополнение к описанным ферментам совсем недавно было показано, что G-белки также модулируют и активность вольтажзависимых ионных каналов. Как видно из таблицы, многие гормоны и нейромедиаторы регулируют как вторичные мессенджеры, так и ионные каналы, активируя один G-белок. В частности, Gt стимулирует как аденилатциклазу, так и некоторые типы Са2+-каналов.
Очевидно, дальнейшее выяснение механизмов прямой и обратной регуляции рецепторных систем сигнальной транедукции является одной из важнейших научных задач будущего.

Представления о молекулярно-клеточных механизмах онкогенной трансформации клеток претерпели значительную эволюцию на протяжении XX века и до настоящего времени [18, 20, 25, 32, 34].
Как указывалось выше, инициирующими этиологическими факторами малигнизации клетки являются разнообразные по природе группы канцерогенов химической, физической, биологической природы, в том числе вирусы, гормоны и генотоксические продукты их метаболизма [13, 26, 63].
Естественно, что при чрезвычайной гетерогенности этиологических факторов неоплазий не могла сформироваться достаточно быстро доминирующая концепция механизмов развития онкогенной трансформации клеток, их активации или промоции опухолевого роста с последующей опухолевой прогрессией. В ранних концепциях канцерогенеза делали акцент на эпигеномных механизмах развития неоплазий, и, безусловно, ряд положений этого направления носит не только исторический характер, но может быть в определенной степени ассоциирован с современными вирусо-генетической и онкогенной теориями канцерогенеза.
Согласно данным ряда исследователей, первичное изменение свойств цитоплазматической мембраны под влиянием канцерогенных углеводородов, онкогенных вирусов является одним из пусковых механизмов последующего изменения генетического аппарата и нарушений регуляции их митотического цикла [108]. Эта концепция канцерогенеза была актуальна в период обнаружения отсутствия контактного ингибирования опухолевых клеток в монослойной культуре.
Как оказалось далее, в механизмах контактного ингибирования клеток важная роль отводится активации мембранной аденилциклазы и увеличению уровня цАМФ, тормозящего митотическую активность клеток. Понижение концентрации цАМФ в мембранах клеток под влиянием различных канцерогенов ведет к неконтролируемой митотической активности. Эта точка зрения имела определенную значимость в понимании пусковых механизмов канцерогенеза, поскольку для многих гормонов, регулирующих метаболизм клеток, их митотическую активность, характерен преимущественно мембранный тип рецепции (АКТГ, СТГ, инсулин, пролактин и др.).
Практически одновременно с мембранной концепцией канцерогенеза создавались митохондриальная и лизосомальная теории развития неоплазий, согласно которым актомиозиновый белок митохондриальных мембран оказывается аномальным у малигнизированных клеток и утрачивает чувствительность к регулирующим влияниям АТФ; при этом гликолитическая реакция опухолевой клетки стимулируется митохондриальными факторами, поступающими постоянно в гиалоплазму, а возрастание концентрации АТФ не подавляет этот процесс.
Одним из классических признаков неоплазий является нарушение регуляции дифференцировки и митотической активности клеток, в связи с чем указанная проблема затрагивается в той или иной форме в разных концепциях [1]. Однако до настоящего времени одной из ведущих концепций канцерогенеза является мутационная теория, согласно которой все канцерогены обладают мутагенной активностью, хотя не все мутагены являются канцерогенами.
Практически все изученные канцерогены индуцируют разрывы фосфодиэстеразных связей в молекуле ДНК. Вначале канцерогены интенсивно связываются с ДНК чувствительных клеток. Обнаружена прямая корреляция между чувствительностью животных и их органов к малигнизирующему действию веществ и степенью их связывания с ДНК [42].
В последующие годы важная роль в развитии онкогенной трансформации клеток и опухолевой прогрессии отведена свободным радикалам. Учитывая значимость индукции избыточных концентраций свободных радикалов в канцерогенезе, необходимо прежде всего остановиться на активации процессов липопероксидации, инициируемой активными формами кислорода (АФК) и в то же время являющейся источником образования значительного количества вторичных эндогенных свободных радикалов [7, 8].
Как известно, активные формы кислорода вступают во взаимодействие с полиненасыщенными жирными кислотами (ПНЖК): линолиевой, линоленовой, арахидоновой – важнейшими компонентами фосфолипидов биологических мембран. Отрыв водорода от молекулы ПНЖК при участии АФК приводит к перемещению двойных связей с образованием гидроперекисей диеновых коньюгатов, которые затем метаболизируются во вторичные (малоновый диальдегид) и третичные продукты липопероксидации [66]. Перекисное окисление липидов затрагивает прежде всего фосфолипиды цитоплазматических мембран клеток, нарушая при этом энергозависимый трансмембранный перенос субстратов, процессы межклеточного взаимодействия. Биологическая активность АФК связана с синтезом простагландинов, лейкотриенов окислительной модификацией белков, нуклеиновых кислот, липидов. Одним из проявлений окислительной модификации белка является инактивация около 240 ферментов, в частности, СОД, ацетил-КоА-гидролазы, каталазы, миелопероксидазы, цитохрома Р450 [22, 66].
Дезинтеграция белка в основном возникает под влиянием гидроксильного радикала, образующегося в организме в процессе реакции взаимодействия супероксида и перекиси водорода с металлами переменной валентности. Объектами окисления в молекуле ДНК под влиянием гидроксильного радикала являются углеводные компоненты, фосфатные группировки, азотистые основания. Наиболее чувствительным к окислительной деструкции азотистым основанием является гуанин, модифицированные формы которого составляют 45 % от общего количества окисленных оснований [83, 95].
Установлено, что чувствительность к фрагментации сахарно-фосфатного остатка ДНК под влиянием АФК оказалось более высокой, чем полипептидного остова белково-пептидных субстанций. Гидроксильный радикал, действуя на ДНК, может отрывать атом водорода от дезоксирибозофосфата, что ведет к его расщеплению и освобождению азотистых оснований. При этом образуются высокотоксичные производные альдегиды.
Данные, опубликованные в последние годы, убедительно свидетельствуют о том, что активные формы кислорода, оксид азота и его производные в сочетании с инфекционными патогенными факторами, бактериями и вирусами, являются ключевыми факторами канцерогенеза [2, 35, 36].
Детальный обзор литературы по этому вопросу приведен в работе Х. Маеда, Т. Акаике (1998). Кислородные радикалы, а также оксид азота могут повреждать ДНК, вызывая мутацию. Мутагенный и канцерогенный эффекты указанных соединений резко возрастают при одномоментной, избыточной продукции, сопровождающейся их взаимодействием с образованием пероксинитрита. Последний участвует в различных внутриклеточных метаболических процессах: нитровании остатков тирозина в белках, подавлении транспорта электронов в митохондриях, в окислении тиоловых соединений. Пероксинитрит является ДНК-расщепляющим агентом. Вышеуказанные химические реакции с участием пероксинитрита могут инициировать апоптоз, мутации, онкогенную трансформацию клеток.
Как указывалось выше, в механизмах индукции канцерогенеза важная роль отводится онкогенным ДНК- и РНК-содержащим вирусам, способным инкорпорировать свою ДНК или ДНК-копию в геном хозяина с последующей возможной онкогенной трансформацией клетки в случае экспрессии протоонкогенов.
Установлено, что РНК-содержащие онкогенные вирусы являются членами семейства ретровирусов, характеризуются наличием липидной оболочки и двух односпиральных РНК, фермента РНК-зависимой ДНК-полимеразы, необходимой для репродукции вируса. Наличие этого фермента обеспечивает обратную транскрипцию вирусной РНК- в ДНК-копию, интегрирующую с геномом клетки [71].
Группа РНК-содержащих вирусов включает следующие разновидности: непатогенную для человека группу вирусов (род А); медленно трансформирующийся вирус гормонзависимой карциномы молочной железы морских свинок и, возможно, человека (род В); дефектные быстро трансформирующиеся и недефектные медленно трансформирующиеся вирусы (род С); род Д – включает вирусы приматов и вирус перевиваемых раковых клеток человека.
ДНК-содержащие онкогенные вирусы подразделяются на следующие семейства:
1. Семейство Poxviridae, содержит, в частности, вирус контагиозного моллюска человека.
2. Семейство Herpes viridae, к которому относится вирус Эпштейн-Барра человеа, вызывающий лимфому Беркитта, цитомегаловирус человека – тип 5.
3. Семейство Adenoviridae – представителями которого являются аденовирусы человека.
4. Семейство Papovaviridae, представителями которого являются вирусы папилломы крыс, хомяков, обезьян, человека.
ДНК-содержащие вирусы внедряют свою ДНК в геном хозяина при участии ферментов эндонуклеаз и липаз, а за счет наличия генов – промоторов – вирусы инициируют транскрипцию генов, следующих за ДНК-вирусами. Последствия внедрения ДНК-вирусов в геном хозяина зависят от зоны инкорнации: интронов, экзонов, протоонкогенов, антионкогенов. Если ДНК-содержащие вирусы встраивают в геном хозяина клетки регуляторы экспрессии протоонкогенов, возможна малигнизация клетки [54].
Механизмы онкогенной трансформации клеток под влиянием ДНК-содержащих вирусов могут быть весьма разнообразны: за счет индукции ранних онкобелков, так называемых Т-антигенов, усиления экспрессии рецепторов экзогенных ростовых факторов. Большие и средние Т-белки ряда ДНК-содержащих вирусов выключают контактное ингибирование пролиферации клеток, препятствуют действию антионкогена р53.
Как известно, вирусо-генетическая теория Л.А. Зильбера явилась основной для формирования современной онкогенной теории канцерогенеза. На смену вирусогенетической теории канцерогенеза пришли теории онкогенов, протоонкогенов и антионкогенов [30, 31, 65, 120].
В настоящее время, очевидно, что в опухолевой трансформации клеток, возникающей под влиянием различных индукторов канцерогенеза, принципиально участвуют следующие категории генов:
1. Онкогены- стимуляторы функций.
2. Гены роста и пролиферации клеток (Myc, Ras, Los, ABL и другие).
3. Антионкогены (потеря функции).
4. Гены, отвечающие за программированную смерть клетки (апоптоз):
– отменяющие программированную смерть: Bcl-2 (стимуляция функций);
– гены смерти клеток – р53 (потеря функции).
Онкогены как специфический химический материал, кодирующий информацию об определенном химическом продукте, впервые были идентифицированы в составе ретровирусов. Геном типичного не трансформирующего ретровируса представляет собой две молекулы односпиральной РНК. Основные гены вируса относятся к трем регионам: gag кодирует структурные белки вирион частицы, env– белки оболочки вириона, ген pol – несет информацию об обратной транскрипции. Последний обеспечивает образование ДНК- копии на матрице РНК-вируса.
Согласно гипотезе онкогенов, гены ретровирусов, попавшие в геном человека в процессе эволюции, переходят по наследству в ряде поколений, проявляют себя в раннем онтогенезе, а затем подавляются внутриклеточными репрессорами. С возрастом под влиянием различных канцерогенов физической, химической, биологической природы возникают экспрессия вирусных онкогенов и усиление продукции ими онкобелков, ответственных за малигнизацию клетки. Онкогенные свойства нетрансформирующих ретровирусов обусловлены наличием в их геноме V-онкогенов, причем большинство из 50 V-онкогенов имеют клеточные прототипы – С-протоонкогены.
Высказывается мысль, что ретровирусы не только могут вносить в определенные позиции клеточного генома V-онкогены, но и способны быть промоторами для усиленной экспрессии протоонкогенов клеток. Считается, что в ходе совместной эволюции ретровирусов и клеток происходят захват клеточных протонкогенов вирусами и их перенос [24].
Развитие теории онкогенов нашло отражение в концепции Темина (1972) о протовирусах, протоонкогенах, согласно которой предсуществующий аналог вируса не является результатом инфекции, а нормальным клеточным геном, необходимым для роста и онтогенеза клеток, причем нормальные клетки не содержат вирусных онкогенов, но зависят от контролируемой экспрессии их клеточных аналогов.
В механизмах развития неоплазий онкогенные ретровирусы играют неоднозначную роль: различают быстро- и медленно-трансформирующие вирусы. Быстротрансформирующие вирусы дефектны по структуре, утратили часть своих поздних репликативных генов и приобрели взамен видоизмененные клеточные гены-V-онкогены, которые и вызывают неопластическую трансформацию при повторной интеграции в клеточный геном. Для полного цикла репликации этим вирусом требуются вирусы-помощники. Клеточные протоонкогены являются прототипами V-онкогенов, консервативными регуляторами клеточной дифференцировки.
Встраивание быстро-трансформирующего реторовируса может либо привести к экспрессии в клетке V-онкогена, либо вирусные промоторы и энхансеры встраиваются рядом с протоонкогенами клетки, вызывая их экспрессию.
Таким образом, встраивание ретровирусов в геном клетки приводит к гиперэкспрессии протоонкогенов, переход их в онкогены с последующей малигнизацией клетки [20, 23, 30, 64].
Что касается механизмов индукции неоплазий химическими канцерогенами с точки зрения современных теорий канцерогенеза – протоонкогенов, онкогенов, антионкогенов, то необходимо остановиться на анализе лишь некоторых работ, посвященных данной проблеме.
Как известно, химические канцерогены, подобно биологическим, способны вызывать развитие мутаций и активацию протоонкогенов [25, 64]. Под влиянием химических канцерогенов возможна онкогенная трансформация в процессе амплификации ДНК. Установлено, что амплификация гена резистентности на фоне воздействия цитостатиков нередко возникает при раке кишечника и является причиной устойчивости неоплазий к химиотерапии. При ряде онкологических заболеваний желудочно-кишечного тракта возникает амплификация онкогенов erbB2, mys, SRS. Индукция развития опухолей нитрозмочевиной связана с амплификацией и активацией N-ras; в опухолях, индуцированных гамма-облучением, активен Ras-H. В ходе химического канцерогенеза отмечено гипометилирование протоонкогена Ras-H, приводящего к развитию генной мутации.
В опухолях, индуцированных химическими канцерогенами, отмечены транскрипции ряда других онкогенов (c-ras и c-mys), связанные с гипометилированием протоонкогена либо его амплификацией. В ходе химического канцерогенеза нарушается зависимость экспрессии c-mys (но не c-ras) от клеточного цикла. Таким образом, многие химические соединения или физические воздействия, а также вирусы могут вызывать мутации ДНК, не летальные для клеток и провоцирующие экспрессию протоонкогенов или депрессию антипротоонкогенов [108]. Последнее приводит к трансформации нормальной клетки в опухолевую.
Эпигенетический механизм канцерогенеза связан с нарушением регуляции клеточного роста, функции клетки и экспрессии генов без повреждения генома. При эпигенетическом канцерогенном эффекте эндогенных или экзогенных канцерогенных факторов возникает инактивация белков-продуктов антипротоонкогенов или активация пострецепторных передатчиков ростовых факторов. Такое воздействие, как правило, не вызывает неоплазии, но усиливает ростовые эффекты, способствует пролиферации мутантного клона и формированию распознаваемой неоплазии. Эффект канцерогенов-мутагенов называют инициирующим, а коканцерогенов – активирующим.
Таким образом, в настоящее время очевидны следующие механизмы активации протоонкогенов:
1) амплификация протоонкогенов, в результате чего резко возрастает их общая активность, что может привести к малигнизации клетки;
2) мутации протоонкогенов, приводящие к их активации, и ингибиция антипротоонкогенов;
3) транслокация протоонкогенов в локус с функционирующим промотором;
4) аддукция промотора рядом с протоонкогеном. В качестве промотора могут выступать ДНК-копии определенных участков онкорнавирусов, а также мобильные генетические структуры, способные перемещаться и встраиваться в различные участки генома.
В геноме человека предполагается наличие около 100 протоонкогенов, выполняющих следующие функции:
1) кодирование ростовых факторов, их рецепторов и пострецепторных передатчиков;
2) кодирование блокаторов запрограммированной гибели клеток, контактного ингибирования пролиферации.
Трансформация протоонкогенов в онкогены приводит к их экспрессии и синтезу онкобелков. При этом онкобелки продуцируются перманентно в увеличенном количестве или в качественно измененном состоянии.
Ниже представлены несколько групп протоонкогенов, антионкогены, и кодируемые ими белки [30, 31, 32].
Читайте также: