
Эпикант это в генетике кратко
Обновлено: 04.07.2024
Принято считать, что человек — венец творения. Однако этому противоречит наличие в наших организмах рудиментов и атавизмов — наследия эволюционного процесса. Для чего они нужны и какую могут нести угрозу?
Говоря об опасности для жизни человека, конечно, уместнее всё же употреблять термин атавизм. В отличие от рудимента, это аномалии развития человека, часть из которых (но не все) при определённых условиях могут стать нозологической формой, то есть болезнью.
Сюда можно отнести сплошной волосяной покров тела, добавочные пары молочных желез, незаращение предсердной перегородки сердца, жаберные мешки, щели (на фоне неполноценно развитого слухового аппарата).
Рудиментом же можно назвать хвостовые позвонки (копчик), ушные мышцы, морганиевы желудочки гортани, зубы мудрости, пирамидальная мышца, червеобразный отросток слепой кишки (аппендикс), эпикантус (третье веко).
Ошибки эволюции. В чём причина?
Несмотря на достаточно явные отличия этих двух групп признаков, у них общая генетическая сущность, общая эволюционная база: органы (признаки), ставшие бесполезными для организма, не утрачиваются в одночасье, а могут сохраняться миллионы лет.Они медленно разрушаются под грузом накапливающихся мутаций.
Кто они, рудименты?
Одним из самых известных рудиментов, пожалуй, является аппендикс и неразрывно связанное и с ним понятие аппендицита, то есть воспаление этого самого червеобразного отростка. Интересно заметить, что в общей хирургической практике операции по поводу аппендицита являются одними из самых частых. Зачастую болезнь таит в себе грозные осложнения в виде перитонита (воспаление ткани покрывающей всю брюшную полость), абсцесса (формирование гнойника брюшной полости).
Особый интерес она представляет в связи с географической и расовой привязанностью. Так, более 60% азиатов имеют эпикантус, в то время как представители негроидной расы, европейцы не имеет его вовсе (почти в 100 процентах).
Таким образом, являясь полезной особенностью у одних групп людей, эпикантус является в полной мере рудиментом — у других. В клинической практике замечено, что этот рудимент более чем в 80% выявляется у больных, страдающих синдромом Дауна.
Болезнь или аномальная особенность?
Геномный импринтинг и эпигенетика. Характеристика
При некоторых болезнях экспрессия фенотипа связана с тем, от кого из родителей унаследован мутантный аллель или аномальная хромосома. Различия экспрессии генов между отцовской и материнской аллелью — результат геномного импринтинга.
Импринтинг — нормальный процесс, вызванный изменениями в хроматине в характерных позициях генома, произошедшими в половых клетках только одного из родителей. Изменения включают ковалентные модификации ДНК, например метилирование цитозина с формированием 5-метилцитозина, или изменения и замены в гистоновых белках хроматина, которые могут влиять на экспрессию генов в конкретной хромосомной области.
Примечательно, что импринтинг влияет на экспрессию генов, а не на первичную последовательность ДНК. Это обратимая форма инактивации гена, а не мутация, таким образом, пример эпигенетигеского эффекта. Эпигенетика — раздел генетики человека и медицинской генетики всевозрастающей важности, изучающий влияние на экспрессию генов и фенотип как у здоровых, так и больных при многих патологиях, включая цитогенетические аномалии, моногенные заболевания и онкологию.
Импринтинг происходит при гаметогенезе, до оплодотворения, и отмечает отдельные гены как исходящие от матери или отца. После зачатия импринтинг регулирует экспрессию генов в пределах импринтируемой области в некоторых или всех соматических тканях эмбриона.
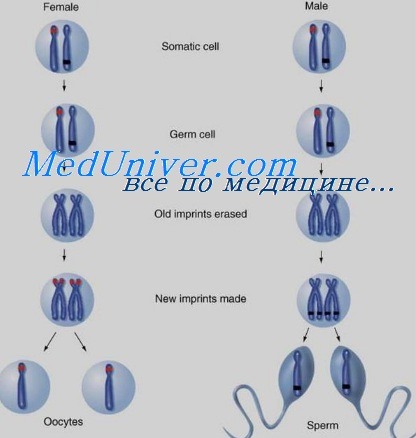
Состояние импринтинга сохраняется и после рождения через сотни клеточных делений, приводя к тому, что в клетке экспрессируется только материнская или отцовская копия гена. В то же время импринтинг обратим: если аллель отцовского происхождения наследуется женщиной, он преобразуется в ее половых клетках, так что она может передать его потомству уже с материнским импринтингом.
Аналогично импринтированный материнский аллель, унаследованный мальчиком, должен преобразоваться в его половых клетках, с тем чтобы стать импринтированным отцовским аллелем у его потомства. Управление данным процессом осуществляют центры импринтинга — специальные элементы ДНК, находящиеся в пределах импринтируемых областей генома. Хотя точный механизм их действия неизвестен, они должны вызывать эпигенетические изменения в хроматине, которые затем распространяются по хромосоме в области импринтинга.
Здесь мы сфокусируемся на влиянии импринтинга в клинической цитогенетике, так как множество его эффектов обнаруживают вследствие хромосомных аномалий.
Подтверждение геномного импринтинга получено для множества хромосом или хромосомных областей всего генома при сравнении фенотипов лиц, несущих ту же цитогенетическую аномалию, затрагивающую материнский или отцовский гомолог. Хотя оценки варьируют, вероятно, что, по крайней мере, несколько десятков, если не сотен генов у человека проявляют эффекты импринтинга.
Некоторые области содержат единственный импринтируемый ген; другие содержат целые группы, в некоторых случаях занимающие более 1 мегабазы вдоль хромосомы, или же множественные гены импринтинга.
Признак импринтированных генов, отличающий их от других аутосомных локусов, — экспресия в тканях только одного аллеля, или материнского, или отцовского. В отличие от этого, в неимпринтируемых локусах всех клеток (т.е. в подавляющем большинстве локусов генома) экспрессируются как материнские, так и отцовские аллели.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Блефарофимоз-птоз-эпикантус-синдром в практике врача-акушера-гинеколога клиники репродукции
Журнал: Проблемы репродукции. 2021;27(1): 67-70



Блефарофимоз-птоз-эпикантус-синдром (blepharophimosis-ptosis-epicanthus syndrome — BPES) — редкий (1:50 000 родов) генетический аутосомно-доминантный синдром, основными проявлениями которого являются блефарофимоз, птоз, эпикантус. Известно два клинических варианта BPES: с нарушением функции яичников, которое проявляется их преждевременным истощением, бесплодием, гипергонадотропной аменореей, и без нарушения функции яичников. Молекулярно-генетической основой синдрома является полиморфизм гена FOXL2, локализованного в длинном плече 3-й хромосомы. Мутация гена FOXL2 описана как одна из генетических форм преждевременной недостаточности яичников. В статье представлено два клинических варианта BPES у молодых женщин, обратившихся в клинику репродукции по поводу планирования беременности. Приведены анамнезы, результаты лабораторных и инструментальных исследований пациенток, а также рассмотрены факторы, затрудняющие диагностику синдрома.
Даты принятия в печать:
Блефарофимоз-птоз-эпикантус-синдром (blepharophimosis-ptosis-epicanthus syndrome — BPES) — генетическое заболевание, которое встречается реже чем 1 случай на 50 000 родов. В связи с этим четкие данные о распространенности синдрома в мире неизвестны. Впервые синдром описал Friedrich August von Ammon (1841 г.) и затем более детально Vignes (1889 г.). BPES характеризуется в первую очередь комплексом офтальмологических симптомов, таких как блефарофимоз, птоз, обратный эпикантус, телекантус. BPES наследуется как аутосомно-доминантное состояние, таким образом, каждый ребенок пациента с BPES имеет 50%-й шанс развития этого заболевания. В 50% случаев BPES носит спорадический характер. Описан единственный случай аутосомно-рецессивной передачи этого синдрома в семье кровных родственников [1—3].
Известны два генетических варианта BPES: BPES I, который встречается чаще, передается мужчиной, а при передаче женщине помимо офтальмологических симптомов приводит к первичной или вторичной аменорее и бесплодию, связанным с преждевременной недостаточностью яичников (ПНЯ) и гипергонадотропным гипогонадизмом, и BPES II, который передается потомству и от мужчины, и от женщины и проявляется комплексом офтальмологических и фациальных симптомов без ПНЯ у женщин [2, 5].
Молекулярно-генетической основой синдрома является полиморфизм гена FOXL2. FOXL2 — это ген, локализованный на длинном плече 3-й хромосомы в позиции 22.3. (Gene ID: 668, updated on 21-Jan-2020). Известен также под названиями BPES, PFRK, POF3, BPES1, PINTO. Описано более 130 полиморфизмов гена FOXL2, связанных с BPES. Продуктом экспрессии этого гена является белок — транскрипционный фактор — Forkhead Box. Изучена регулирующая роль этого фактора в пролиферации и дифференцировке различных тканей, в том числе мышц века и клеток гранулезы яичника. Помимо полиморфизма непосредственно гена FOXL2 описаны хромосомные мутации 3-й хромосомы, которые затрагивают ген FOXL2 и поэтому могут проявляться другими симптомами кроме тех, что типичны для BPES [2, 4—7].
Описание клинических случаев
В период с 2013 по 2020 г. в клинике, специализирующейся на лечении бесплодия методами вспомогательных репродуктивных технологий (ВРТ), наблюдались две пациентки.
Из анамнеза жизни: физическое и интеллектуальное развитие в соответствии с возрастом, начало первой менструации в 13 лет, менструации регулярные, по 5—6 дней, через 28—30 дней.
При общем осмотре обращает на себя внимание резкое сужение глазной щели, нависание верхнего века, необычно широко расположенные глаза. При общении пациентка отклоняет голову назад, чтобы лучше видеть собеседника. Носит корректирующие очки с 12 лет (VD=VS= –2,50) без прогрессирования миопии по настоящий момент (февраль 2020 г.). Индекс массы тела нормальный, телосложение обычное, кожный покров и развитие подкожной жировой клетчатки обычные, соответствуют полу. При гинекологическом осмотре в зеркалах и бимануально — без особенностей.
Результаты лабораторного обследования в 2014 г.: антимюллеров гормон (АМГ) — 3,4 нг/мл, фолликулостимулирующий гормон (ФСГ) — 6,3 мМЕ/мл, лютеинизирующий гормон (ЛГ) — 7,4 мМЕ/мл, эстрадиол — 130 пмоль/л, пролактин — 340 мМЕ/л.
Проведено лечение бесплодия с использованием донорских ооцитов и сперматозоидов мужа. С 2015 по 2018 г. проведено три переноса размороженных эмбрионов в полость матки. В 2015 г. и 2018 г. наступала беременность, но в сроке 7—8 нед диагностирована неразвивающаяся беременность по типу анэмбрионии. Выполнено медикаментозное прерывание беременности. В 2016 г. результат переноса отрицательный.
Пациентка прекратила лечение и вновь обратилась в декабре 2019 г. На момент обращения уровень АМГ — 2,7 нг/мл, уровни ФСГ, ЛГ в пределах референсных значений, уровень ФСГ без тенденции к росту.
Пациентка A. На момент обращения в 2019 г. возраст 28 лет, в браке, планирует беременность. Жалобы на нерегулярные менструации, бесплодие в течение 5 лет.
Анамнез заболевания: менструации нерегулярные с 22 лет, задержки по 3—4 мес. После назначения и последующей отмены комбинированных оральных контрацептивов менструации приходят, проба с гестагенами положительная. За последние 5 лет у пациентки 7 раз диагностирована биохимическая беременность. В связи с задержкой менструаций самостоятельно сдавала кровь на исследование содержания хорионического гонадотропина человека бета (β-ХГЧ). Результаты в пределах 18—77 мМЕ/мл (подтверждены документально). Затем при задержке не более 10 дней от ожидаемого срока менструаций спонтанно начиналось кровотечение обычной обильности и продолжительности. В период повышения уровня β-ХГЧ самостоятельно обращалась с целью выполнения ультразвукового исследования. Плодное яйцо в полости матки ни разу не визуализировалось. В связи с жалобами обращалась в женскую консультацию по месту жительства. Из архива результатов обследования пациентки в 2017 г. (возраст 26 лет): уровень АМГ — 0,9 нг/мл, уровень ФСГ — 11,3 мМЕ/мл. По данным ультразвукового исследования — мультифолликулярные яичники. Остальные результаты лабораторных и инструментальных исследований без особенностей. Несмотря на низкий уровень АМГ, пациентке проведено оперативное лечение по поводу синдрома поликистозных яичников. Выполнены лапароскопия, дриллинг яичников. Спустя 4 мес после оперативного лечения уровень АМГ составил 0,4 нг/мл. В период после оперативного лечения дважды наступала биохимическая беременность. С 2018 г. сохраняется нерегулярный характер менструаций.
Из анамнеза жизни: развивалась в соответствии с возрастом, начало первой менструации в 13 лет, менструации нерегулярные и скудные с 19 лет, через 3—4 мес. Помимо этого, в возрасте 5 лет проведено оперативное лечение по поводу блефарофимоза, эпикантуса и птоза. Подобных офтальмологических и фациальных симптомов у других членов семьи не было.
При общем осмотре: пациентка использует бесконтактную коррекцию зрения (очки), обращает на себя внимание сужение глазной щели, умеренное нависание верхнего века. Индекс массы тела нормальный, телосложение, кожный покров и развитие подкожной жировой клетчатки обычные и соответствуют полу.
При гинекологическом осмотре в зеркалах и бимануально — без особенностей. Данные ультразвукового исследования органов малого таза на 5-й день цикла (основные сведения): матка — 52×36×44 мм, миометрий без особенностей, шейка матки — 30 мм, М-эхо — 3 мм, соответствует дню цикла. Яичник справа — 36×36×31 мм, визуализируется более 20 фолликулов диаметром от 2 до 9 мм, яичник слева — 35×37×29 мм, визуализируется более 20 фолликулов диаметром от 2 до 7 мм. Обращает на себя внимание несоответствие уровня АМГ ультразвуковой картине яичников.
Результаты лабораторного обследования в 2019 г. (после менструальноподобной реакции на отмену приема гестагенов): содержание АМГ — 0,1 нг/мл, ФСГ — 30,2 мМЕ/мл, ЛГ — 10,4 мМЕ/мл, эстрадиол — 32 пмоль/л, пролактин — 435 мМЕ/л. Пациентке проведено цитогенетическое исследование — вариант 46, ХХ, 22 pstk+[15], интерпретирован как вариант нормы.
После консультации пациентка продолжает попытки забеременеть самостоятельно, а при неудаче собирается прибегнуть к ВРТ с использованием донорских ооцитов.
Пациентка L., очевидно, относится к клиническому типу BPES II [2], поскольку в данном случае не наблюдается клинических, лабораторных или инструментальных признаков снижения овариального резерва. Пациентка A. — это пример варианта BPES I, при котором наряду с комплексом офтальмологических и фациальных симптомов наблюдаются признаки снижения овариального резерва — снижение уровня АМГ менее 1 нг/мл, повышение уровня ФСГ более 30 мМЕ/мл. В случае с пациенткой А. помимо генетической причины снижения овариального резерва имело место ятрогенное вмешательство. Наличие ультразвуковой картины, напоминающей мультифолликулярную структуру яичников, нарушение менструального цикла и бесплодие, по-видимому, заставили принять решение о проведении оперативного лечения в объеме дриллинга яичников. Интересным является тот факт, что при ультразвуковом исследовании в 2020 г. на фоне уровня АМГ 0,1 нг/мл в яичнике действительно визуализируется большое количество фолликулов (более 20). Это ввело в заблуждение в отношении высокого овариального резерва. Единственное, что обращает на себя внимание, — неправильность формы этих фолликулов.
Среди пациентов с BPES (с наличием или отсутствием преждевременного истощения яичников) описаны различные варианты полиморфизма гена FOXL2, в том числе BPES без полиморфизма гена FOXL2, а также полиморфизм гена FOXL2 без развития BPES [7, 8].
Необходимо понимать, что ценность цитогенетического исследования в диагностике и подтверждении BPES очень ограниченна, за исключением редчайших случаев, когда ген FOXL2 оказывается вовлечен в хромосомную патологию всей третьей пары хромосом.
Отдельный интерес представляет цитогенетическая особенность, выявленная у пациентки А., а именно вариант 46, ХХ, 22 pstk+. Увеличение длины спутничных нитей является нередкой разновидностью хромосомного полимофризма. Молекулярной основой хромосомного полиморфизма является изменение содержания в хромосоме дезоксирибонуклеиновой кислоты (ДНК) с многократно повторяющимися нуклеотидными последовательностями. В случае пациентки А. обнаружен хромосомный полиморфизм в виде увеличения длины спутничных нитей 22-й хромосомы. Изучив литературу, мы обнаружили противоречивые данные о значении этого вида полиморфизма в реализации репродуктивной функции и эффективности ВРТ [9—12]. По-видимому, увеличение длины спутничных нитей 22-й хромосомы является дополнительной особенностью пациентки, никак не связанной с наличием у нее BPES.
Заключение
Женщин с комплексом офтальмологических и фациальных симптомов, специфических для блефарофимоз-птоз-эпикантус-синдрома, следует относить к группе риска по преждевременному истощению яичников. Тактику консультирования таких пациенток следует выбирать, исходя из возможного сокращения репродуктивного периода жизни этих женщин.
Принято считать, что человек — венец творения. Однако этому противоречит наличие в наших организмах рудиментов и атавизмов — наследия эволюционного процесса. Для чего они нужны и какую могут нести угрозу?
Говоря об опасности для жизни человека, конечно, уместнее всё же употреблять термин атавизм. В отличие от рудимента, это аномалии развития человека, часть из которых (но не все) при определённых условиях могут стать нозологической формой, то есть болезнью.
Сюда можно отнести сплошной волосяной покров тела, добавочные пары молочных желез, незаращение предсердной перегородки сердца, жаберные мешки, щели (на фоне неполноценно развитого слухового аппарата).
Рудиментом же можно назвать хвостовые позвонки (копчик), ушные мышцы, морганиевы желудочки гортани, зубы мудрости, пирамидальная мышца, червеобразный отросток слепой кишки (аппендикс), эпикантус (третье веко).
Ошибки эволюции. В чём причина?
Несмотря на достаточно явные отличия этих двух групп признаков, у них общая генетическая сущность, общая эволюционная база: органы (признаки), ставшие бесполезными для организма, не утрачиваются в одночасье, а могут сохраняться миллионы лет.Они медленно разрушаются под грузом накапливающихся мутаций.
Кто они, рудименты?
Одним из самых известных рудиментов, пожалуй, является аппендикс и неразрывно связанное и с ним понятие аппендицита, то есть воспаление этого самого червеобразного отростка. Интересно заметить, что в общей хирургической практике операции по поводу аппендицита являются одними из самых частых. Зачастую болезнь таит в себе грозные осложнения в виде перитонита (воспаление ткани покрывающей всю брюшную полость), абсцесса (формирование гнойника брюшной полости).
Особый интерес она представляет в связи с географической и расовой привязанностью. Так, более 60% азиатов имеют эпикантус, в то время как представители негроидной расы, европейцы не имеет его вовсе (почти в 100 процентах).
Таким образом, являясь полезной особенностью у одних групп людей, эпикантус является в полной мере рудиментом — у других. В клинической практике замечено, что этот рудимент более чем в 80% выявляется у больных, страдающих синдромом Дауна.
Болезнь или аномальная особенность?
Читайте также: