
Этапы секвенирования с использованием метода терминаторов кратко опишите
Обновлено: 02.07.2024
Секвенирование ДНК – это процесс определения последовательности нуклеотидов в ДНК молекула, каждый организм ДНК состоит из уникальной последовательности нуклеотидов. Определение последовательности может помочь ученым сравнить ДНК между организмами, что может помочь показать, как эти организмы связаны.
Обзор секвенирования ДНК
Это означает, что, упорядочив отрезок ДНК, можно будет узнать порядок, в котором четыре нуклеотид основы – аденин, гуанин, цитозин и тимин – происходят в этом нуклеиновая кислота молекулы.
Необходимость секвенирования ДНК впервые стала очевидной из теории Фрэнсиса Крика о том, что последовательность нуклеотидов в молекуле ДНК непосредственно влияет на аминокислотные последовательности белков. В то время считалось, что полностью секвенированный геном приведет к квантовому скачку в понимании биохимия клеток и организмов.
Пример секвенирования ДНК
Хотя секвенирование ДНК раньше занимало годы, теперь это можно сделать за часы. Далее, первая полная последовательность человеческой ДНК заняла около 3 миллиардов долларов. Теперь некоторые компании упорядочат весь ваш геном менее чем за 1000 долларов. Самые продвинутые тесты будут анализировать каждый нуклеотид в вашем геноме. Тем не менее, многие компании сейчас предлагают однонуклеотидный полиморфизм тесты.
Эти тесты фокусируются на отдельных нуклеотидах в генах, которые могут обозначать определенные генетические варианты. Эти SNP, как они известны, связаны с определенными условиями и могут помочь предсказать, как ваши гены могут влиять на вашу жизнь. Некоторые SNP связаны с различными заболеваниями, в то время как другие связаны с вашим метаболизмом и тем, как ваш организм перерабатывает питательные вещества. Были найдены тысячи различных корреляций, и можно использовать секвенирование ДНК, чтобы выяснить, как ваш геном влияет на вашу жизнь.
Методы секвенирования ДНК
Существует два основных типа секвенирования ДНК. Более старый, классический метод обрыва цепи также называется методом Сэнгера. Новые методы, которые могут быстро обрабатывать большое количество молекул ДНК, в совокупности называются методами высокопроизводительного секвенирования (HTS) или методами секвенирования следующего поколения (NGS).
Секвенирование
Метод Сэнгера опирается на праймер, который связывается с молекулой денатурированной ДНК и инициирует синтез одноцепочечного полинуклеотида в присутствии фермента ДНК-полимеразы, используя денатурированную ДНК в качестве матрицы. В большинстве случаев фермент катализирует добавление нуклеотида. Ковалентная связь поэтому образует между 3 ‘атомом углерода дезоксирибоза молекула сахара в одном нуклеотиде и 5 ‘атом углерода в следующем. Это изображение ниже показывает, как формируется эта связь.

В самых ранних попытках использования метода Сэнгера молекулу ДНК сначала амплифицировали, используя меченый праймер, а затем разделяли на четыре пробирки, каждая из которых имела только один тип ddNTP. То есть каждая реакционная смесь будет иметь только один тип модифицированного нуклеотида, который может вызвать обрыв цепи. После того, как четыре реакции были завершены, смесь молекул ДНК, созданная путем обрыва цепи, подвергалась электрофорезу в полиакриламидном геле и разделялась в соответствии с их длиной.
Со временем этот метод был модифицирован таким образом, чтобы каждый ddNTP имел различную флуоресцентную метку. Праймер больше не был источником радиоактивной метки или флуоресцентной метки. Этот метод, также известный как секвенирование терминатора красителя, использовал четыре красителя с неперекрывающимися спектрами излучения, по одному для каждого ddNTP.


Высокая пропускная способность
Секвенирование Сэнгера продолжает быть полезным для определения последовательностей относительно длинных участков ДНК, особенно в небольших объемах. Однако, это может стать дорогим и трудоемким, когда большое количество молекул должно быть быстро секвенировано. Как ни странно, хотя традиционный метод терминатора красителя полезен, когда молекула ДНК длиннее, высокопроизводительные методы стали более широко использоваться, особенно когда необходимо секвенировать целые геномы.
Есть три основных изменения по сравнению с методом Сэнгера. Первой была разработка клетка система для клонирования фрагментов ДНК. Традиционно участок ДНК, который нужно было секвенировать, сначала клонировали в прокариот плазмида и усиливается в бактерии перед извлечением и очисткой. Технологии секвенирования с высокой пропускной способностью или секвенирования следующего поколения больше не полагались на эту трудоемкую и длительную процедуру.
Появление HTS значительно расширило приложения для геномики. Секвенирование ДНК стало неотъемлемой частью фундаментальной науки, трансляционных исследований, медицинской диагностики и криминалистики.
Использование секвенирования ДНК
С другой стороны, HTS позволяет использовать секвенирование ДНК для понимания однонуклеотидных полиморфизмов – среди самых распространенных типов генетическая изменчивость в пределах Население, Это становится важным в эволюционной биологии, а также в обнаружении мутировавших генов, которые могут привести к болезни. Например, вариации последовательности в образцах аденокарциномы легкого позволили обнаружить редкие мутации, связанные с заболеванием. Сайты связывания хроматина для специфических ядерных белков также могут быть точно идентифицированы с использованием этих методов.
В целом, секвенирование ДНК становится неотъемлемой частью многих различных приложений.
диагностика
Секвенирование генома особенно полезно для выявления причин редких генетических нарушений. В то время как более 7800 заболеваний связаны с паттерном менделевского наследования, менее 4000 из этих болезней были окончательно связаны с конкретным геном или мутация, Ранний анализ экзон -геном или экзом, состоящий из всех экспрессируемых генов организма, показал многообещающую роль в выявлении причинных аллелей для многих наследственных заболеваний. В одном конкретном случае секвенирование генома ребенка, страдающего тяжелой формой воспалительного заболевания кишечника, связало заболевание с мутацией в гене, связанном с воспалением – XIAP. В то время как у пациента первоначально были множественные симптомы, указывающие на иммунодефицит, по результатам секвенирования ДНК была рекомендована пересадка костного мозга. Ребенок впоследствии излечился от недуга.
Кроме того, HTS был важным игроком в развитии лучшего понимания опухолей и раковых заболеваний. Понимание генетической основы опухоли или рака позволяет врачам иметь в своем комплекте дополнительный инструмент для принятия диагностических решений. Атлас генома рака и Международный консорциум по геному рака секвенировали большое количество опухолей и продемонстрировали, что эти разрастания могут значительно различаться с точки зрения их мутационного ландшафта. Это также позволило лучше понять, какие варианты лечения являются идеальными для каждого пациента. Например, секвенирование генома рака молочной железы идентифицировало два гена – BRCA1 и BRCA2 – чьи патогенные варианты оказывают огромное влияние на вероятность развития рака молочной железы. Люди с некоторыми патогенными аллелями даже предпочитают проводить профилактические операции, такие как двойная мастэктомия.
Молекулярная биология
Секвенирование ДНК в настоящее время является неотъемлемой частью большинства биологических лабораторий. Он используется для проверки результатов упражнений на клонирование, чтобы понять влияние определенных генов. HTS-технологии используются для изучения вариаций генетического состава плазмид, бактерий, дрожжей, нематод или даже млекопитающих, используемых в лабораторных экспериментах. Например, клеточная линия, полученная из рака молочной железы ткань называется HeLa, используется во многих лабораториях по всему миру и ранее считался надежной клеточной линией, представляющей ткани молочной железы человека. Недавние результаты секвенирования продемонстрировали большие различия в геноме клеток HeLa из разных источников, тем самым уменьшая их полезность в клеточной и молекулярной биологии.
Секвенирование ДНК дает представление о регуляторных элементах в геноме каждой клетки и об изменениях их активности у разных типов клеток и индивидуумов. Например, определенный ген может быть постоянно выключен в некоторых тканях, в то время как конститутивно экспрессируется в других. Точно так же люди с восприимчивостью к определенному заболеванию могут регулировать ген иначе, чем те, кто обладает иммунитетом. Эти различия в регуляторных областях ДНК могут быть продемонстрированы с помощью секвенирования и могут дать представление о основе для фенотип.
Последние достижения даже позволили отдельным лабораториям изучать структурные изменения в геноме человека – мероприятии, которое потребовало глобального сотрудничества два десятилетия назад.
Криминалистика
Возможность использовать низкие концентрации ДНК для получения надежных результатов секвенирования была чрезвычайно полезна для судебно-медицинских экспертов. В частности, потенциал для последовательности каждой ДНК в образце является привлекательным, особенно потому, что место преступления часто содержит генетический материал от нескольких людей. HTS медленно внедряется во многих криминалистических лабораториях для идентификации человека. Кроме того, последние достижения позволяют судмедэксперту упорядочить экзом человека после смерти, особенно для определения причины смерти. Например, смерть от отравления покажет изменения экзома в пораженных органах. С другой стороны, секвенирование ДНК может также определить, что умерший имел ранее существовавшую генетическую болезнь или предрасположенность. Проблемы в этой области включают разработку чрезвычайно надежного программного обеспечения для анализа, тем более что результаты HTS не могут быть проверены вручную.


Метод пиросеквенирования основан на детекции активности фермента ДНК-полимеразы с другим хемилюминесцентным ферментом. Последовательность подачи реагентов в реакционную смесь, которые дают хемилюминесцентный сигнал, позволяет определить последовательность анализируемого участка ДНК.
Секвенирование ДНК в последние десятилетия превратилось из узкой области, которой занималось небольшое число ученых, в одну из самых стремительно развивающихся технологий. Рост производительности и падение стоимости даже опережают закон Мура, и, из-за большой конкуренции на рынке и огромного спроса, развитие и дальше будет идти высокими темпами. Кроме того, развитие секвенирования привело к такому же буму в биоинформатике и коренным образом изменило биологию, и, постепенно, также основательно меняет медицину.

По катом я подробнее рассказываю, как это делают.
Что такое ДНК
Для начала, чтобы понимать сам процесс, немного необходимой теории.
ДНК — это полимерная цепь, состоящая из мономеров четырех типов, называемых нуклеотидами, последовательность которых и кодирует информацию об организме. Иначе говоря, ДНК можно представить как текст, написанный четырехбуквенным алфавитом. ДНК — молекула, состоящая из двух цепочек, и, хотя, последовательность нуклеотидов у них разная, последовательность одной цепочки можно однозначно восстановить, если известна последовательность другой. Поэтому цепочки называют комплементарными. (англ. Complement – дополнение) Это свойство используется при копировании клетки, когда цепочки ДНК расплетаются, и, на каждой, как на матрице, синтезируется вторая, и каждая из двух дочерних клеток получает свою двуцепочечную ДНК. Вся последовательность ДНК организма называется геномом. Например, геном человека состоит из 46 хромосом.
Несмотря на большое количество разнообразных, как экспериментальных, так и устаревших методов, мейнстримовые коммерческие методы довольно похожи, и, чтобы не делать оговорки каждый раз, сразу скажу, что речь дальше будет идти именно об этих мейнстримовых методах.
Как это выглядит в общем
Перед описанием технологии секвенирования, для интуитивного понимания, проведу следующую аналогию: стопку одинаковых газет взрывают так, что они разлетаются на небольшие кусочки с отрывками текста, а, затем, каждый из этих кусочков читают и, из этих прочтений восстанавливают текст первоначальной газеты.
Чтобы секвенировать ДНК, сначала ее выделяют из исследуемого образца, затем режут на небольшие фрагменты случайным образом, фрагменты называются ридами. От каждого рида оставляют по одной цепочке, и на этой цепочке, как на матрице, синтезируют вторую, причем, тип каждого следующего присоединяющегося нуклеотида как-то детектируют. Таким образом, записывая последовательность присоединившихся нуклеотидов, восстанавливают их последовательность в каждом риде. Затем, из последовательностей ридов с помощью компьютерных программ реконструируют геном.
Важный момент. Суммарная длина ридов должна многократно превышать длину исследуемой ДНК. Делается это потому, что, когда ДНК выделяют из образца, и когда ее режут, часть ее теряется, так что никто не гарантирует, что каждый ее участок попадет хотя бы в один рид. Поэтому, чтобы каждый участок гарантированно был бы прочтен, ДНК берут с большим запасом. Кроме того, при секвенировании возможны ошибки, и, чтобы более надежно прочитать ДНК, каждый ее участок следует прочитать несколько раз.

ДНК разрезают на риды, которые читают, и из них восстанавливают первоначальную последовательность
Такая методика используется не от хорошей жизни. Она добавляет множество трудностей, и, если бы исследователи могли взять и прочитать за раз целую последовательность генома, то они были бы счастливы, однако, это на данный момент невозможно.
У этого есть 2 причины. Первая — это ошибки, происходящие при чтении каждого нуклеотида. Они постепенно накапливаются, и, каждый следующий нуклеотид читается хуже предыдущего, и, в какой-то момент качество чтения настолько снижается, что дальше продолжать процесс бессмысленно. У разных методов секвенирования длина рида, которы они могут хорошо прочитать, составляет порядка десятков или сотен нуклеотидов. Вторая заключается в том, что ДНК — это очень длинная молекула, и, при скрупулезном чтении каждой буквы друг за дружкой, секвенирование заняло бы неприлично много времени, а в данном случае этот процесс легко распараллеливается, и можно одновременно читать миллионы и миллиарды ридов.

Illumina
Такая схема в общих чертах описывает все популярные методики секвенирования. Различаются они лишь методами детекции присоединившихся нуклеотидов при синтезе, и методикой подготовки материала.
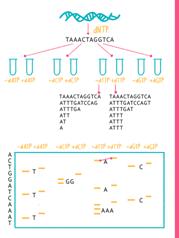
На сегодняшний день самым распространенным является метод, который используется в секвенаторах компании Illumina. В этом методе сначала множество различных ридов прикрепляется к стеклянной пластине. Затем, с каждого рида делают множество копий на поверхности пластины так, чтобы на каждом ее небольшом участке располагались лишь одинаковые копии. Это делается для того, чтобы при последующем секвенировании получать сигнал не от одиночной молекулы, а от группы одинаковых молекул, располагающихся рядом. Так и сигнал легче считывать, и надежность считывания увеличивается. Эти молекулы являются одноцепочечными ДНК, и на них в процессе секвенирования синтезируются комплементарные цепи. Реакцию синтеза проводят следующим образом: К началу каждой молекулы присоединяется по одному нуклеотиду. Этот нуклеотид химически блокирован так, что после его присоединения синтез дальше не идет. Кроме того, к нему присоединена метка, которая под действием лазера люминесцирует. Причем, для каждого типа нуклеотидов цвет люминесценции разный. После присоединения нуклеотида пластину освещают лазером и фотокамера фиксирует цвета, которыми люминесцирует пластина. После этого блокировку снимают, метку также снимают, и присоединяют таким же образом следующий нуклеотид. Последовательность световых сигналов на каждом участке пластины в компьютере переводится в последовательность нуклеотидов, и, на выходе получается файл, содержащий последовательности ридов.

Секвенирование по методу Illumina
1 — геномная ДНК 2 — разрезается на риды 3 — к ридам прикрепляются адаптеры, с помощью которых они приклеиваются на 4 — пластину 5 — размножение ридов на пластине 6 — засовывам в секвенатор и 7 — секвенируем
Сборка и аннотирование генома
Если геномы близких организмов раньше не секвенировались, то из ридов, затем, с помощью программ, пытаются собрать единую последовательность нуклеотидов. Риды частично перекрываются, и, с помощью этих перекрытий пытаются выстроить единую последовательность. Здесь есть множество моментов, которые существенно осложняют дело. Например, можно загрязнить образец, и программа будет пытаться выстроить одну последовательность из ДНК разных организмов. Секвенатор может ошибиться при чтении рида, или неверно связать два места в геноме, потому что они очень похожи. На самом деле, сложностей так много, что всех тут не перечислишь. И, некоторые из них настолько сложно поддаются устранению, что, даже геном человека, самый важный и широко исследуемый геном, все еще не секвенирован до конца.

риды и внизу последовательность генома, которая реконструирована на их основе
Если геном другого организма этого вида уже секвенировался, то его используют, для сборки. Так как геномы разных организмов одного вида различаются лишь незначительно, то для каждого рида находят место на секвенированном геноме, к которому он ближе всего, и на основе этого генома собирают новый.
![]()
Геномика: постановка задачи и методы секвенирования

Сергей Николенко, кандидат физико-математических наук, старший научный сотрудник лаборатории вычислительной биологии Санкт-Петербургского Академического Университета в серии статей говорит о некоторых задачах биоинформатики, связанных со сборкой и анализом геномов, делая акцент на математической, комбинаторной постановке задачи. В данном, вводном, тексте речь идет о том, как выглядят входные данные для сборки геномов и как их получают.
Как выглядит молекула ДНК?

Рисунок из Википедии
Что такое секвенирование?
Клонирование происходит либо просто выращиванием клеток в чашке Петри, либо (в случаях, когда это было бы слишком медленно или по каким-то причинам не получилось бы) при помощи так называемой полимеразной цепной реакции. В кратком и неточном изложении работает она примерно так: сначала ДНК денатурируют, т.е. разрушают водородные связи, получая отдельные нити. Затем к ДНК присоединяют так называемые праймеры; это короткие участки ДНК, к которым может присоединиться ДНК-полимераза – соединение, которое, собственно, и занимается копированием (репликацией) нити ДНК.

Рисунок из Википедии
На следующем этапе полимераза копирует ДНК, после чего процесс можно повторять: после новой денатурации отдельных нитей будет уже вдвое больше, на третьем цикле – вчетверо, и так далее.
Секвенирование по Сэнгеру
Первым методом секвенирования, который учёные сумели применить для обработки целых геномов (в том числе генома человека), стало секвенирование по Сэнгеру (Sanger sequencing). Смысл таков: участок ДНК клонируется, после чего полученная смесь делится на четыре части. Каждая часть помещается в активную среду, где присутствуют:
ATGCAGAACAGACGATCAGCGACACTTTA (образец)
AT
ATGCAGAACAGACGAT
ATGCAGAACAGACGATCAGCGACACT
ATGCAGAACAGACGATCAGCGACACTT
ATGCAGAACAGACGATCAGCGACACTTT
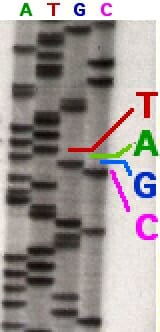
Очевидно, что эта последовательность начинается с А (т.к. самый лёгкий префикс, из одной буквы, заканчивается на A); дальше идёт C, дальше опять A, и так далее. В результате можно прочесть исходный участок: ATGCAGAACA.

Рисунок из Википедии
Видно, что (в идеальном случае) можно просто прочесть последовательность нуклеотидов от самого лёгкого префикса (т.е. префикса из одной буквы) к самому тяжёлому.
Результаты и ошибки сэнгеровского секвенирования
На выходе из сэнгеровского секвенатора получаются короткие участки ДНК, так называемые риды (reads). Для биоинформатики принципиальны две вещи: во-первых, какой длины получаются риды, во-вторых, какие в них могут быть ошибки и как часто (разумеется, на свете нет ничего идеального).
Сэнгеровские риды по этим критериям очень хороши: получаются риды длиной около тысячи нуклеотидов, причём качество начинает заметно падать только после 700-800 нуклеотидов. Сам процесс секвенирования по Сэнгеру, с которым мы познакомились в предыдущем разделе, предопределяет и эффект падения качества (труднее отличить молекулу массой 700 от молекулы массой 701, чем массу 5 от массы 6), и другой неприятный эффект – если в геноме встречается длинная последовательность из одной и той же буквы (…AAAAAAAA…), трудно бывает точно определить, какой она длины – все промежуточные массы попадут в одну и ту же пробирку, некоторые из них могут не встретиться, некоторые слиться друг с другом и т.д. Но всё же сэнгеровское секвенирование даёт отличные результаты с достаточно длинными ридами, которые потом относительно легко собирать. О том, как это делается, мы будем говорить в последующих текстах.
Именно при помощи сэнгеровского секвенирования был впервые расшифрован геном человека. Секвенирование по Сэнгеру применяется и сегодня, но его всё активнее вытесняют другие методы, и применяется оно всё реже. Кому же и почему оно уступило свои позиции?
Секвенаторы второго поколения: Illumina
Современные секвенаторы – это так называемые секвенаторы второго поколения (SGS, second generation sequencing). В них участки ДНК по-прежнему многократно клонируются, но процесс чтения устроен не так, как у Сэнгера. Существует много разных методов, отличающихся довольно существенно, поэтому мы рассмотрим только один из них, один из самых популярных на сегодня – секвенирование по методу Solexa (ныне Illumina; в смене названия не нужно искать глубокий смысл, просто одна компания купила другую).
Процесс секвенирования Illumina проиллюстрирован на рисунке; кроме того, можно посмотреть один из нескольких существующих видеороликов с анимацией этого процесса – в данном случае, действительно, лучше один раз увидеть, чем сто раз прочесть текст. Однако краткие комментарии тоже пригодятся; вот как происходит процесс секвенирования по методу Illumina.
В результате на каждом цикле мы прочитываем одновременно очень большое число нуклеотидов из разных последовательностей. Но за это приходится платить тем, что участки ДНК, которые мы можем прочесть, оказываются гораздо короче, чем в случае секвенирования по Сэнгеру – риды Illumina обычно получаются длиной около 100 нуклеотидов.
Парные риды и постановка задачи
Итак, теперь мы можем формально поставить задачу сборки геномов. Она звучит так: по большому числу подстрок небольшой длины восстановить исходную длинную строку в алфавите из букв A, C, G, T. В случае секвенирования по методу Illumina – по большому числу пар коротких подстрок, разделённых в исходной строке приблизительно известным расстоянием. Поставив эту задачу, мы можем забыть про биологию, химию и медицину – перед нами чисто алгоритмическая задача. Однако, прежде чем перейти к математике, сделаем ещё несколько замечаний.
Ошибки и показатели качества в секвенаторах второго поколения
Как мы уже знаем, секвенирование всегда содержит ошибки. В секвенаторах Illumina и аналогичных ошибки, как правило, происходят на фазе, когда нужно распознать помеченные нуклеотиды, т.е. понять, каким цветом и с какой силой светятся кластеры из многократно клонированных участков ДНК. На рисунке – типичный пример такой фотографии, порождённой секвенатором Illumina.

Рисунок с сайта medicine.yale.edu
Проблема здесь заключается в том, что из-за неидеальности остальных этапов процесса кластеры никогда не светятся только одним цветом; это всегда смесь всех четырёх цветов с той или иной интенсивностью. Нужно выделить наиболее интенсивную компоненту и оценить, насколько вероятна ошибка в этой букве; эта задача называется base calling (распознавание нуклеотидов). Base calling – это целая наука, в подробности которой мы сейчас вдаваться не будем.
Для нас сейчас важно, что в результате каждому нуклеотиду каждого рида секвенатор ставит в соответствие вероятность того, что этот нуклеотид был распознан правильно. Эти вероятности тоже можно использовать при сборке, и секвенаторы выдают их вместе с собственно ридами.
В итоге типичный рид в так называемом fastq-формате, стандартном для секвенаторов второго поколения, выглядит примерно так:
Первая и третья строки содержат имя рида; вторая строка – сама последовательность нуклеотидов. Обратим внимание, что среди букв A, C, G, T встречаются и буквы N – это значит, что секвенатор не смог однозначно определить, какой здесь был нуклеотид, и сдался. А четвёртая строка кодирует, в логарифмическом масштабе, вероятности того, что тот или иной нуклеотид распознан правильно; например, H здесь соответствует вероятности ошибки около одной десятитысячной. Как правило, качество ухудшается к концу рида; в нашем примере, как видите, хвост рида и вовсе не удалось сколь-нибудь надёжно прочитать.
Другие методы секвенирования
Хотя мы подробнее всего рассмотрели секвенатор Illumina (Solexa), на самом деле на этом методе свет клином не сошёлся. Есть и другие секвенаторы второго поколения, с другими свойствами.
Пиросеквенирование (pyrosequencing) основано на хемилюминесцентных сигналах, которые подают специально модифицированные нуклеотиды, когда соединяются с комплементарным нуклеотидом в прочитываемой нити ДНК; на этом принципе работает, например, секвенатор 454 от 454 Life Sciences.
Недавно появившийся метод ионного полупроводникового секвенирования (на нём основан секвенатор IonTorrent) вместо всего этого просто детектирует соединения (ионы), которые выделяются при присоединении нового нуклеотида к нити ДНК. Это позволяет радикально сократить время и стоимость получаемых ридов, хотя процент ошибок становится больше, и больше становится ошибок в фрагментах из повторяющейся одной буквы.
Человеческая мысль не стоит на месте: методы секвенирования постоянно улучшаются. Однако практически все современные методы выдают относительно короткие риды, от 100 до 400 нуклеотидов; в этом цикле мы будем в основном говорить о том, как собирать именно короткие риды.
Sanger или Illumina?
Человеческий геном был впервые собран на сэнгеровских секвенаторах, причём алгоритмическая сторона того проекта была проработана гораздо меньше, чем сейчас, десять лет спустя. Алгоритмы, которыми собирали первый человеческий геном, значительно проще тех, о которых мы будем говорить в дальнейшем. Однако первый геном всё-таки собрали; может быть, весь алгоритмический прогресс – это никому не нужный миф, и вполне хватило бы старых программ?
На таком уровне становится важной и цена алгоритмической стороны вопроса. Чтобы сборка геномов не занимала дольше и не стоила дороже, чем само их секвенирование, нужно разработать очень быстрые алгоритмы для решения задачи сборки. Об этом пойдет речь в следующей статье.
Читайте также:
- Инструкция о порядке обращения с документированной служебной информацией ограниченного доступа в доу
- Квазиреальная ситуация начальная школа пример
- Социально философские взгляды вольтера и монтескье дух времени и дух закона кратко
- Темы консультаций в доу по развитию речи
- Костюм на хэллоуин для девушек своими руками в домашних условиях в школу