
Синдром мартина белл доклад
Обновлено: 18.05.2024
Это заболевание впервые было описано J. Martin, J. Bell в 1943 г. В 1969 г. H. Lubs обнаружил хромосомный маркер — хромосому X с пробелом в субтеломерном участке длинного плеча Xq27.3. Отсюда основное название синдрома — синдром фрагильной (ломкой) Х-хромосомы. В 1991 г. удалось показать, что при этом синдроме множественные повторы последовательности CGG в Xq27.3 являются причиной локального гиперметилирования и повреждения синтеза белка [Verk ег К. et al., 1991]. В общей популяции здоровые индивидуумы имеют от 5 до 50 таких тринуклеотидных повторов, носители же мутантного гена FMR1 — от 50 до 200 повторов. Если же число повторов превышает 200, то появляется полный фенотип синдрома ломкой хромосомы X, и метилированный FMR1 ген не продуцирует белок [Devys D. e t al., 1993]. Функции белка FMRP неизвестны. Предполагается, что его отсутствие сказывается на процессах развития ЦНС. В мозге этот белок присутствует во всех нейронах и более представлен в сером веществе. В период эмбрионального развития концентрация FMRP особенно велика в базальном гигантоклеточном ядре, которое является поставщиком холинергических нейронов для лимбической системы.
Существует широкий спектр нарушений у больных с синдромом ломкой Х-хромосомы. Лица женского пола с полной мутацией более сохранны, чем лица мужского пола. У них в 30 % случаев не отмечается умственной отсталости.
Частота встречаемости 1:2000 у лиц мужского пола и от 2,5 до 6 случаев на 100 детей с УМО [Herbst D. S., Miller J. R., 1980; Hagerman R. et al., 1988].
Патогенез заболевания остается невыясненным.
Больным с Х-ФРА свойствен специфический физический фенотип, определяемый следующими стигмами дизонтогенеза. Дети имеют череп долихоцефалической формы, удлиненное лицо с выступающим лбом и прогенией. Ушные раковины оттопырены и увеличены. Нос с широким основанием, кончик его клювовидный, опущены углы рта, нередко встречаются высокое небо, подслизистые расщелины неба и язычка. Средняя часть лица уплощена. Пальцы рук удлинены, стопы плоские. Наблюдаются макроорхизм (после пубертатного периода), гипотония.
Повышена эластичность кожных покровов, аорты, сердечных клапанов. Отмечается слабость связочного аппарата коленных и голеностопных суставов [Козлова С. И. и др., 1987; Маринчева Г. С. и др., 1988; Денисова Л. В., 1988; Gillberg Ch. , 1995].
Наблюдается отставание в умственном и речевом развитии. Когнитивные функции недостаточны; IQ варьирует от 70 до 35, в большинстве случаев ниже 50 [Hagerman R. J., Suverman A., 1991]. Речь с небольшим запасом слов. Часты и более высокие достижения в вербальных задачах. Большинство девочек имеют IQ от нормального уровня и ниже нормы — 80—90. Отмечаются дизлексия, дискалькулия.
Бедность словарного запаса с годами становится очевидной, появляются неравномерность темпа речи, однообразность тембра, неуправляемая громкость. Речевой поток с ускоренным выпаливанием отдельных слов сменяется затуханием громкости речи с возникновением нечеткости в произношении звуков, что создает похожесть ее на эгоцентрическую речь у детей с детским аутизмом. При этом само общение нарушается и начинает походить на аутистическое с отказом от социальных контактов со сверстниками и родными.
Наличие в клинической картине больных с Х-ФРА таких переходов в активности и реагировании только отдаленно напоминает симптом переслаивания функций у больных с синдромом аутизма Каннера, но не идентично ему. У больных с Х-ФРА нет истинного смешения ранних и более поздних функций, а наблюдается более целостное реагирование, как бы соответствующее то более раннему, то более зрелому возрасту. Дезинтеграции в разных функциональных системах не возникает. Следует отметить, что имеются единичные случаи с Х-ФРА, неотличимые от синдрома раннего детского аутизма.
На последующих возрастных этапах в личностной структуре больных с Х-ФРА сохраняются черты сенситивности, повышенной чувствительности, смущаемости с быстрым отказом от любого общения, с избеганием глазного контакта. Возрастает количество привычных моторных стереотипии в виде потирания ладоней рук, потряхивания кистями рук. В речи отмечаются стереотипные повторы слов, эхолалии слов, фраз. На фоне появляющегося интереса к окружающим детям контакты с ними практически не формируются, общение затруднено, но не достигает глубины классического аутизма.
С годами деятельность становится монотоннее, все более упрощаются интересы, побуждения становятся резко недостаточными. Больные обращаются к одним и тем же игровым сюжетам, игровые фантазии отличаются крайней обедненностью сюжетов, из года в год поведение повторяется, как клише. Углубляются трудности перехода к новым формам деятельности. Поведение становится примитивным, в нем нет парадоксальности и вычурности.
Эмоциональное развитие, привязанность к родным соответствуют уровню психического развития.
Наличие примитивных моторных стереотипии и бедность побудительных мотивов являются помехой в усложнении и формировании моторных навыков, необходимых в самообслуживании. Особое внимание обращает на себя нарастающая торпидность в мышлении, действиях, поведении. При этом легко возникают реакции раздражительности, протеста, а также невротические реакции в ответ на психогенные воздействия.
С увеличением возраста больных все грубее становятся когнитивные проблемы. Уровень IQ не возрастает, умственная отсталость (без диссоциированности) не смягчается, а достигает устойчивой стабилизации. Структура интеллектуального дефекта носит равномерный характер.
По данным R. Hodapp и соавт. (1990), в пубертате возможна остановка в развитии, с регрессом и значительным снижением IQ, с сохранением робости, избегания взгляда.
Свойственные этим детям спады и подъемы активности смягчаются. Аутизм проявляется в сужении круга общения, стереотипной, обедненной деятельности, в недостаточности речевого общения. Уровень социализации соответствует тяжести умственного недоразвития, в большинстве своем эти дети нуждаются в уходе и надзоре в течение всего периода жизни, многие дети заканчивают свою жизнь в учреждениях собеса.
При проведении дифференциального диагноза с аутизмом шизофренического спектра следует опираться на отсутствие диссоциации в умственном развитии, особых манеризмов в моторике, смягчение аутизма с возрастом ребенка, частично на фоне лечения и реабилитации.
Дифференциальный диагноз помогает проводить цитогенетический анализ, показывающий Х-ломкую хромосому в 2—70 % случаев. Аутизм при Х-ФРА в ряде случаев носит нерезко выраженный характер и не ведет к дезинтеграции в деятельности.
Терапия. Считается, что специфического лечения при Х-ФРА нет [Hagerman R., Silverman A., 1991]. Многие авторы предлагают пользоваться стимуляторами [Hagemian R. J., M urphy M. A., Wittenberger M. D., 1988].
Для уточнения диагноза необходимо проводить исследование на молекулярном уровне.
Таким образом, аутистический круг расстройств при Х-ФРА возникает позже, после 1—2 лет жизни ребенка; глубина его меньше, чем при классическом аутизме, моторные стереотипии проще и охватывают более зрелые моторные формулы, эмоциональная сфера никогда не бывает такой скудной, глазная реакция более сформирована или вовсе не повреждена, нет синдрома переслаивания зрелых и менее зрелых невытесненных функций. Все перечисленные особенности аутистическиподобных расстройств при Х-ФРА и послужили основанием к тому, чтобы их определять аутистическиподобными, а не аутистическими симптомами.
Однако и аутистическиподобный комплекс симптомов при Х-ФРА несет в себе основные признаки аутизма: отрешенность, стереотипность в деятельности и движениях, диссоциацию в развитии. Возможно, что аутистическиподобные синдромы при Х-ФРА связаны с поражением не вполне идентичных с синдромом Каннера структур мозга, но, по-видимому, все же очень близких и определенным образом взаимосвязанных, что подлежит дальнейшему изучению.
Приводим клиническое наблюдение больного с Х-ФРА
Ребенок М., 9 лет, от 1-й беременности, первых родов. Раннее психомоторное развитие близко к возрастной норме. После полугода стал отставать в становлении речи, крупных моторных актов. С годами нарастали возбудимость, раздражительность, моторное беспокойство. С детьми не играл. В обособленной игре (в одиночку) игрушки обнюхивал, вертел, лизал. Тактильный контакт с матерью отвергал. Настроение было снижено, часто без внешних на то причин кричал, бил себя, появлялась тенденция к разрушительству. С годами возрастала неуправляемость.
Психический статус при поступлении. Общение отвергает. Негативен. Осмотру подчиняется с трудом. На вопросы отвечает невнятно. Речь — одно-, двусловная, с аграмматизмами. Речевая тональность, тембр изменены. Словарный запас обеднен. Речь легко становится невнятной, затухшей, близкой к эгоцентрической. Периодами истощается и уходит от общения, в беспокойстве перемещается по кабинету, оставляя без внимания все попытки общения с ним. Через несколько минут вновь удается на короткий срок привлечь его внимание. Энергетический потенциал флюктуирующий и очень низкий, любая деятельность вначале отвергается ребенком, после активного многократного побуждения к ней удается получить один-два ответа в плане вопросов, а затем вновь наступают отторжение контакта, моторное беспокойство с раздражительностью. В кистях рук сохраняются примитивные атетозоподобные стереотипии. В целом поведение полевое, с временами наступающим возбуждением, самоагрессией и разрушительными тенденциями.
В психическом статусе на первый план выступает тяжелое умственное недоразвитие с аутистическими тенденциями, которые позволили диагностировать детский аутизм. Структура аутизма была столь глубока и очевидна, что на этом этапе состояние приближалось к детскому аутизму Каннера.
Логопедическое обследование. Задержка психоречевого развития, аутистический синдром.
Неврологический статус. ЧМН — без видимой патологии. Ходит самостоятельно. Тонкая моторика кистей рук не сформирована. Сухожильные рефлексы не изменены. Координация не нарушена. Патологических знаков нет. Болевая чувствительность сохранена. Тазовые функции не нарушены.
Соматический статус. Долихоцефальный череп, удлиненное лицо, массивный подбородок. Большие оттопыренные уши, прогнатизм, высокое небо, нос с широким основанием и клювовидным кончиком. Кисти и стопы увеличены. Кожа гиперэластичная, растяжима. Суставы с повышенной разгибаемостью. Пролапс митрального клапана. Другой патологии со стороны внутренних органов не отмечено.
Катамнез: 9 лет 10 мес. После проведения 1-го курса лечения церебролизином по методу Осипенко — Скворцова смягчились моторная расторможенность и аутистическая отрешенность. Улучшились ориентация в окружающем, память, увеличился словарный запас, стал усваивать новые знания. В связи с заболеванием сестры проведено цитогенетическое обследование и кариотипирование детей и матери. Обнаружены 46 XY, Х-ФРА. У пробанда и сибса при цитогенетическом исследовании обнаружена ломкая Х-хромосома.
Катамнез: 10 лет 9 мес. После трех курсов терапии по методу Осипенко — Скворцова в НТЦ состояние резко улучшилось. Расширилось общение с окружающими, смягчилась аутизация. Появилось стремление к игровой деятельности, способен к наблюдению и подражанию в ней. Начал обучаться в школе. Выучил буквы. Сидит на уроках самостоятельно, без матери.
Полностью исчезли моторное возбуждение, беспричинный страх, бесцельное беспокойство кататоноподобного характера. В кистях рук не стало манеризмов. Осознает временные события. Стал пользоваться личными местоимениями, восстановилось осознание себя, использует личные местоимения по отношению к себе. Исчезло симбиотическое отношение к матери. Помогает ей в уходе за сестрой. Оживились эмоции.
Логопедическое обследование. Улучшилась связанная речь, стал употреблять более сложные по звуковому составу слова.
Заключение. В настоящее время в статусе на первый план выступает умственное недоразвитие со значительным нивелированием аутистической симптоматики, снятием кататоноподобного возбуждения и страхов. При первом осмотре ребенка в возрасте 9 лет статус определяли проявления аутизма, стереотипии в пальцах рук, симптомы тождества, особые речевые расстройства в форме контаминации, незавершенности фраз, их разорванности, негативизм, фобии, моторное возбуждение с агрессией, которые служили основанием для диагностики аутизма.
Периодами состояние утяжелялось, сопровождаясь регрессом приобретенных навыков, присоединением фобического синдрома, оживлением моторного возбуждения с агрессивными тенденциями. Наличие этих симптомов, казалось бы, только подтверждало диагностику классического аутизма.
Положительная динамика состояния с почти полным купированием аутистических расстройств, обнаружение при цитогенетическом обследовании ломкой Х-хромосомы послужили основанием к постановке диагноза: УМО, Х-ФРА, аутистическиподобный синдром.
Настоящий случай представляет несомненный интерес в отношении как динамики состояния при лечении новым методом по Осипенко — Скворцову, так и нивелирования собственно аутистических симптомов при Х-ФРА, а также уменьшения проявлений умственного недоразвития. Следующее наблюдение.
Ребенок М., 5 лет 1 мес, от 2-й беременности, протекавшей без патологии. Роды в срок, нормальные .
Психомоторное развитие. Голову держит с 1 мес, сидит с 6 мес, ходит с 13 мес. Первые слова с 1,5 года; фразовой речи нет.
К 3 годам в поведении стали обращать на себя внимание задержка в становлении речи, отказ от общения, однообразная стереотипная, примитивная игра, особые постукивания пальцами рук по предметам, сосание пальцев рук, периодические двигательные возбуждения с негативностью.
В 4,5 года поступила в НТЦ профилактики и лечения детской неврологической инвалидности.
Психический статус при поступлении. Отрешена. Глазной реакции нет, на звук, зов реакция отставленная. Обследованию сопротивляется. Речь эгоцентрическая, понимание элементарной фразы присутствует, ответы иногда носят противоположный характер. Настроение индифферентно-раздражительное. На показ игровых предметов, сладостей положительно не реагирует, напротив, следует отказная, протестная реакция, стремление к уходу от источника раздражения. В целом поведение однообразное, бездеятельное. В пальцах рук стереотипные манеризмы. Сон не расстроен, аппетит вялый, избирательный выбор пищи.
Неврологический статус. ЧМН без патологии. Двигательная сфера: ходит самостоятельно, расторможена. Сухожильные рефлексы живые, равномерные. Патологических знаков нет. Координация не нарушена. Болевая чувствительность сохранена. Дисфазия.
ЭЭГ: -ритм 7 кол/с, регулярный, 1000 мкВ. Эпикомплексы отсутствуют; -ритм незначительный, -волны — множественные.
Изменения на ЭЭГ резидуально-органического генеза свидетельствуют о дисфункции диэнцефально-стволовых структур.
Диагноз: атипичный РДА, сдвиг в развитии между 1 — 1,5—3 годами. Отставание в умственном развитии на 2— 3,5 возрастных порядка.
После двух курсов терапии в НТЦ состояние ребенка улучшилось. Увеличился словарный запас, стала иногда пользоваться двусловными предложениями. Речь не использует в качестве средства коммуникации. По инструкции не работает. Аутохтонная деятельность расширилась: реагирует на детские передачи, показанные по телевизору, иногда наблюдает за игрою детей. Стала периодами выполнять просьбы матери. Можно сказать, что отрешение стало менее выраженным, больше осознает ситуацию.
ЭЭГ-изменения резидуально-органического генеза; в сравнении с предыдущей ЭЭГ установлено некоторое улучшение параметров корковой активности; -ритм — 8 кол/с.
Логопедическое обследование. Активизировался лепет, легче идет на занятия к логопеду.
Цитогенетическое исследование. При цитогенетическом исследовании и детей, и матери: обнаружена ломкость участка Xq27.3, позволившая подтвердить диагноз Х-ФРА.
Таким образом, на основании обследования и наблюдения можно подтвердить диагноз: синдром Мартина — Белл, или Х-сцепленная умственная отсталость с ломкой Х-хромосомой (Х-ФРА), аутистическиподобный синдром.
Интерес в данном случае представляет поздняя диагностика Х-ФРА, коморбидность с аутистическиподобным синдромом, большое сходство болезненного состояния с процессуальным аутизмом, значительное улучшение состояния после терапии.
Еще в начале нашего века клинические исследования выявили значительное преобладание лиц мужского пола среди больных олигофренией. Впервые в 1934 г. J. Martin и J. Bell была описана семья, где умственная отсталость наследовалась по сцепленному с полом типу. В 2-х поколениях большой семьи из Англии насчитывалось 11 умственно отсталых мужчин и 2 женщины с более легким интеллектуальным дефектом. Далее таких семей описывалось все больше, в 1969 г. H. Lubs, проводя цитогенетическое обследование умственно отсталого мальчика из семьи с рецессивным, сцепленным с полом типом наследования, выявил у него вторичную перетяжку на длинном плече Х-хромосомы в области 27-28.
Частота встречаемости
Среди новорожденных мальчиков частота заболевания составляет от 1 на 1000 до 1 на 2000. Таким образом, частота распространенности этой формы умственной отсталости среди новорожденных лишь немного уступает распространенности синдрома Дауна.
Выявленная цитогенетическая аномалия относится к феномену так называемой фрагильности (fragility - ломкость, хрупкость). При данной патологии важно то, что ломкость затрагивает всегда один и тот же участок Х-хромосомы, где локализован мутантный ген. Ни патогенетическая роль выявленного феномена, ни механизм его связи с мутантным геном до сего времени не выяснены. При цитогенетическом исследовании обнаружили не во всех метафазных клетках. Было выявлено, что обнаружение его зависит от определенных условий среды, таких как отсутствие фолиевой кислоты.
Вопрос о наследовании синдрома Мартина-Белл изучен недостаточно, лишь в 1/3 случаев оно характеризуется как типичное Х-сцепленное рецессивного типа. Это означает, что заболевание проявляется исключительно у лиц мужского пола. Генотип мужчины устанавливается по его фенотипу: если он болен, то гемизиготен по аномальному гену, если здоров, то свободен от него. Женщины-носительницы мутантного гена, как правило, фенотипически здоровы. Среди мужчин-родственников матери по женской линии нередко встречаются больные с тем же заболеванием. Среди здоровых членов семьи по женской линии преобладают женщины, также как и среди здоровых сибсов пробанда. Однако в большем числе случаев (2/3) наследование синдрома носит характер нерегулярного сцепленного с Х-хромосомой, а именно получены строгие доказательства возможности передачи маркерной хромосомы здоровыми мужчинами (облигатными носителями), в потомстве дочерей которых отмечаются случаи рождения больных сыновей. Для ген, обуславливающего синдром, определена неполная его пенетрантность у мужчин (79%). Кроме того, женщины-носительницы в ряде случаев (35%) имеют умственную отсталость от легкой до тяжелой ее форм. Хотя матери и дочери мужчин-носителей гена ломкости Х-хромосомы сходны по фенотипу, экспрессия этого гена в их потомстве различна: пенетрантность умственной отсталости выше в потомстве интеллектуально нормальных дочерей мужчин-носителей, чем в потомстве интеллектуально нормальных матерей мужчин-носителей. Эта ситуация, как и передача синдрома здоровыми мужчинами, хорошо объясняется аутосомной супрессорной системой, которая обеспечивает неполную пенетрантность гена.
Ген-супрессор подавляет проявление мутантного гена у облигатного носителя-мужчины с нормальным интеллектом. У его дочерей вследствие рекомбинационных процессов ген-супрессор из системы генотипа исчезает, что является условием для появления мутантного гена у их сыновей. Предположение об аутосомном гене-супрессоре в противоположность супрессору, находящемуся в одной группе сцепления (Х-хромосома) допускает большую частоту их рекомбинаций (независимое комбинирование). Это повышает вероятность такой комбинации генотипа в потомстве, когда мутантный ген входит в систему генотипа без гена-супрессора и формирует соответствующий синдром у носителя.
Существует гипотеза о ломкости Х-хромосомы как о некотором премутационном состоянии локуса Хq27.3, сопровождающимся аномалиями рекомбинации в этом районе. В последующих поколениях они способствуют возникновению мутантного аллеля, обусловливающего синдром Мартина-Белл. Авторы при помощи своей гипотезы объясняют уже упомянутые особенности родословных с нерегулярным наследованием синдрома, согласно которым обеспечивается передача мутантного гена нормальными мужчинами, причем их гетерозиготные дочери не имеют умственной отсталости, у них либо нет ломкого участка, либо количество таких клеток мало. В следующем поколении треть женщин-гетерозигот имеют умственную отсталость и около 29% с ломкой Х-хромосомой. Эти женщины имеют больных сыновей.
Женщины-носительницы мутантного гена могут быть умственно отсталыми, что свидетельствует в пользу того, что мутантный ген не является полностью рецессивным. Пенетрантность умственной отсталости у гетерозигот у равна 30%. Степень умственной отсталости варьирует от легкой до тяжелой, но чаще женщины-носительницы мутантного гена психически нормальны. Изменчивость интеллекта у женщин-носительниц аномального гена ряд авторов объясняют различиями в инактивации ломкой Х-хромосомы в соответствии с гипотезой J. Lejeune, т.е. у женщин с нормальным интеллектом рано реплицирующейся (генетически активной) чаще бывает нормальная Х-хромосома, тогда как у женщин со сниженным интеллектом - ломкая Х-хромосома.
Было отмечено, что средняя масса тела пробандов при рождении часто повышена - от 3,5 до 4 кг.
Первым в качестве одного из наиболее выраженных соматических симптомов у подростков и взрослых больных отметили макроорхизм при отсутствии изменений эндокринной функции.
Прежде всего определенные признаки были отмечены в строении лица и тела больных : большая голова с высоким и широким лбом, длинное лицо с увеличенным подбородком, несколько уплощенная средняя часть лица, тупой, слегка клювовидно загнутый кончик носа. Уши большие, иногда оттопыренные, низко расположенные. Кисти и стопы широкие, дистальные фаланги пальцев также широкие, суставы имеют повышенную подвижность. Кожа нередко гиперэластична. Часто встречаются светлоокрашенные радужные оболочки, светлые волосы. Вместе с тем вариабельность фенотипа очень широкая.
Таким образом, выявленные системные соматические изменения затрагивают связочный аппарат, хрящ, кожу, костную систему. Это дает основание предполагать, что в патологический процесс вовлекается соединительная ткань.
По данным практически всех исследователей, неврологическая симптоматика при данном синдроме характеризуется некоторыми слабо выраженными и неспецифическими признаками, которые часто встречаются при умственной отсталости у детей вообще. Чаще всего выявляется мышечная гипотония и дискоординация движений. Кроме того, отмечаются экстрапирамидные (стереотипные гримасы), пирамидные и глазодвигательные нарушения.
Ведущим психопатологическим нарушением является интеллектуальное недоразвитие , которое отмечается всеми исследователями как постоянный синдром. По данным литературы, степень умственной отсталости различна. Вместе с тем есть работы, где описываются больные, имеющие пограничную умственную отсталость и даже с интеллектом, соответствующим норме.
Было обращено внимание на своеобразную речь этих пациентов. Позднее речь пациентов стали называть “повторяющейся, персеверативной, бормочущей”. К ее особенностям относится ускоренный темп и выраженные эхолалии и персеверации. Она является весьма характерной именно для этой патологии и может иметь определенное диагностическое значение.
Часто выявляются разнообразные нарушения поведения : синдром двигательной расторможенности, выраженная аффективная возбудимость, агрессивность.
В качестве одной из частых психопатологических особенностей отмечена шизофреноподобная симптоматика, включающая в себя подпрыгивания, похлопывания руками, повороты вокруг своей оси, встряхивание кистями, “манежный” бег, разнообразные гримасы, монотонное хныканье.
Помимо умственной отсталости, чаще всего у больных отмечаются проявления раннего детского аутизма (отсутствие потребности в контактах с окружающими, отгороженность от внешнего мира, слабость эмоционального реагирования по отношению к близким , недостаточность реакций на зрительные и слуховые раздражители, приверженность к сохранению неизменности окружающего, боязнь всего нового, однообразность поведения со склонностью к стереотипным примитивным движениям, разнообразные расстройства речи, непереносимость взгляда в глаза, взгляд “мимо” и “сквозь” людей.
Методы диагностики
1. Клинический. К минимальным диагностическим признакам относятся: умеренная или глубокая умственная отсталость (97,5%), большие оттопыренные ушные раковины (62%), выступающий лоб и массивный подбородок, макроорхизм (68,4%), характерная речь (41,4%).
2. Цитогенетический. Существует стандартный метод изучения культуры лимфоцитов периферической крови больных. Культуру получают на среде 199 (дефицитной по фолиевой кислоте) с добавлением 15% сыворотки крупного рогатого скота. Далее через 72 часа ее фиксируют и окрашивают с помощью трипсин-гимза. Анализируют на стадии метафазы, учитывают число клеток с ломкой Х-хромосомой на 100 изученных клеток.
3. Были описаны характерные для синдрома изменения на ЭЭГ: отсутствие a-ритма, значительное усиление q-диапазона, преобладание нерегулярной q- и D-активности в затылочных зонах коры, в центральных, лобных и теменных отделах полушарий отмечено доминирование высокоамплитудного q-ритма.
4. При анализе дерматоглифических данных с помощью разработанной дискриминантной классификационной функции их диагностическая значимость составляет 80%.
Подходы к лечению
В связи с тем, что ломкость Х-хромосомы можно выявить лишь в среде, обедненной фолатами, возникло предположение о роли дефицита фолиевой кислоты в патогенезе самого заболевания. Это привело к попыткам лечить умственную отсталость введение фолатов.
Эффект от лечения был более выражен у детей, чем у взрослых. В целом все авторы отмечают, что у больных при лечении препаратами фолиевой кислоты не происходит улучшения в интеллектуальном развитии, но улучшается поведение: уменьшаются гиперактивность, агрессивность, повышается внимание, моторная координация, улучшается качественная и количественная сторона речи.
Было предложено применять психостимуляторы. Авторы отмечают неплохие результаты, особенно при лечении метилфенидатом. Дети становились более внимательными, сосредоточенными, у них уменьшалась двигательная активность.
В 1981 г. ломкую Х-хромосому впервые обнаружили в культуре амниотических клеток. Более точные результаты получаются при исследовании крови плода, чем при биопсии плаценты.
Профилактика
Риск рождения больного ребенка существенно различается в зависимости от того, унаследована ли мутация или же она возникла de novo. В последнем случае риск рождения больного ребенка крайне мал. “Свежесть” мутации можно предположить в том случае, если среди родственников нет больных мужчин и не отмечены случаи умственной отсталости у женщин, при этом у родственников тест на ломкую Х-хромосому отрицателен. Но, учитывая неполную пенетрантность гена, точно доказать спорадический случай невозможно, поэтому несмотря на единичный случай заболевания в семье, не следует утверждать, что мутация возникла de novo. Исходя из этого ребенка считают сегрегантом, а мать - гетерозиготой по аномальному гену, и следующую беременность рекомендуют вести под контролем ломкости Х-хромосомы у плода.
1. Куприянова Т.А. “Синдром Мартина-Белл (умственная отсталость с ломкой Х-хромосомой)”// Журнал невропатологии и психиатрии - 1991. - т.91 - вып.8 - с. 115-125.
2. С.И. Козлова, Н.С. Демикова, Е. Семанова, О.Е. Блинникова “Наследственные синдромы и медико-генетическое консультирование” - М.: Практика, 1996г.
3. Зукин В.Д. “Клинико-генетический анализ синдрома “ломкой” Х-хромосомы”: Автореф.дис. канд. мед. наук/ Киев. ин-т усоверш. врачей. - Киев, 1991 г.
4. Куприянова Т.А., Горькова С.А., Маринчева Г.С. “Клиническая и цитогенетическая диагностика синдрома Мартина-Белл”// Журнал невропатологии и психиатрии - 1991. - т.91 - вып.8 - с. 54-57.
Синдром Мартина-Белл – это наследственная болезнь, которая характеризуется стойким интеллектуальным снижением, расстройствами аутистического спектра и специфическими фенотипическими особенностями. Ключевой симптом – недостаточность познавательных функций. Отмечается гиперактивность, дефицит коммуникативных способностей, замкнутость. Лицо удлиненное, ушные раковины большие, лоб выступающий, кончик носа загнутый. Диагностика основывается на клинико-анамнестических данных и результатах биогенетического анализа. Лечение симптоматическое, включает использование медикаментов и психолого-педагогическую коррекцию.
МКБ-10
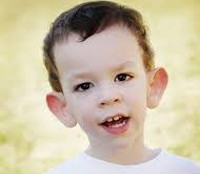
Общие сведения
Причины
Синдром Мартина-Белл является результатом дефекта гена FMR1, расположенного в X-хромосоме. Наследование происходит по доминантному сцепленному с полом типу с неполной пенетрантностью. У мужчин присутствует одна X-хромосома, поэтому мутантный аллель всегда провоцирует болезнь. У женщин есть две половые хромосомы типа X: одна активная, другая – резервная, инактивированная. Таким образом, при наличии мутации в одном из двух генов FMR1 заболевание проявляется или нет в зависимости от активности измененной хромосомы. Мужчины с ломкой хромосомой X не могут передать ее сыновьям, но передают всем дочерям, которые либо болеют, либо остаются здоровыми носителями мутации. Женщины с дефектной хромосомой передают ее детям обоих полов с вероятностью 50%. Наследование синдрома учащается от поколения к поколению, этот феномен называется парадоксом Шермана.
Патогенез
При секвенировании FMR1-гена было выявлено, что основой симптоматики и цитогенетически определяемой ломкости хромосомы X является многократное увеличение количества единичных тринуклеотидов ЦГГ. Это приводит к подавлению транскрипции и последующему недостаточному производству белка FMR1, ответственного за развитие центральной нервной системы, а именно – за формирование аксонов и синапсов, появление и усложнение нейронных связей, успешность процессов обучения и запоминания.
Симптомы
Дети рождаются с увеличенной массой тела, в среднем – 3,5-4 кг. Первыми обращают на себя внимание фенотипические особенности младенцев. Характерен макроорхизм – увеличение яичек без эндокринного заболевания. Окружность головы больше нормы или соответствует ее верхним границам. Лоб высокий и широкий, лицо вытянутое с уплощенной средней частью. Нос имеет слегка клювовидный загиб, ушные раковины крупные, располагаются низко. Суставы отличаются хорошей подвижностью, кости кистей и стоп широкие. Кожа зачастую гиперэластичная, волосы и радужные оболочки глаз светлого оттенка. Фенотипические признаки могут быть выражены по-разному, от одного-двух едва определяемых до полного комплекса.
Ключевое клиническое проявление заболевания – умственная отсталость. Стойкое интеллектуальное снижение проявляется слабым развитием сложных форм мышления и памяти. Пациентам недоступно понимание абстрактно-логических высказываний и явлений, использование категорий, установление аналогий. Сравнение, анализ и обобщение могут осуществляться на простом уровне, например, в конкретных бытовых ситуациях. Словарный запас обеднен. У многих мальчиков IQ равен 40-50 баллам, реже достигает 70-79. Относительно сохранна номинативная речь и зрительное восприятие. У девочек когнитивное снижение менее выраженное, соответствует легкой степени олигофрении или пограничному уровню интеллектуального развития.
Другой типичный симптом заболевания – своеобразие речи. Она ускоренная, сбивчивая, изобилует повторами, эхолалиями и персеверациями. Аутистические расстройства представлены трудностями коммуникации и поведенческими нарушениями. Дети часто проявляют агрессивность и замкнутость при попытке установления контакта. В тяжелых случаях развивается мутизм – полное отсутствие речи как средства общения. В поведении преобладает двигательная расторможенность, гиперактивность, стереотипии, самопровреждения. Пациенты избегают смотреть в глаза, не допускают прикосновений, но по сравнению с больными аутизмом интерес к общению присутствует. Стереотипные движения включают хлопки руками, прыжки, вращения вокруг своей оси, встряхивания руками, бег по кругу, гримасничанье и однообразное хныканье. Имеются трудности планирования и контроля поведения, переключения внимания и пространственной координации.
Неврологические симптомы неспецифичны. Определяется легкое снижение мышечного тонуса, двигательная дискоординация. Недостаточное развитие мелкой моторики затрудняет освоение письма, некоторых игровых и бытовых навыков (сборки конструктора, рисования, шитья и др.). У части больных имеются глазодвигательные нарушения, усиление сухожильных рефлексов, экстрапирамидные паракинезы, например, зажмуривание глаз, нахмуривание бровей, гримасничанье. При тяжелых формах синдрома возникают эпилептические припадки. У 25% пациенток с премутационным состоянием развивается первичная недостаточность яичников.
Диагностика
При выраженных фенотипических изменениях заболевание может быть обнаружено с первых месяцев жизни ребенка – неонатологи и врачи-педиатры обращают внимание на увеличенные размеры яичек и характерные особенности лица. В иных случаях подозрение на умственную отсталость возникает в возрасте от полугода до 2-3 лет. В этот период прослеживается отставание умственного развития, поведенческие и речевые нарушения. Дифференциальная диагностика нацелена на исключение РАС, в частности раннего детского аутизма, а также умственной отсталости другого происхождения (не связанной с ломкостью хромосомы Х). Обследование проводится психиатрами, неврологами и врачами-генетиками, включает:
- Клинический опрос, осмотр. В беседе с ребенком на первый план выходит снижение интеллекта, гиперактивность и расторможенность поведения, нарушение коммуникативных навыков. Уровень психического развития не соответствует возрасту, методики исследования интеллекта выявляют олигофрению (IQ – 40-79 баллов). Внешне наблюдаются характерные фенотипические признаки, при неврологическом осмотре выявляется мышечный гипотонус, усиленные сухожильные рефлексы, паракинезы.
- Генеалогический анализ. В отличие от других форм олигофрении при синдроме Мартина-Белл прослеживается наследственная передача болезни. Как правило, у пациента имеются родственники с данным заболеванием, чаще – мужчины (дед, дядя, брат). Иногда признаки легкого интеллектуального снижения обнаруживаются у матери, но диагноз у нее часто не установлен (не подтвержден).
- Генетическое исследование. В лабораторных условиях исследуется строение ДНК: определяется количество ЦГГ-повторов и статус метилирования. Применяется ПЦР и цитогенетический метод. Диагноз подтверждается, если количество триплетных повторов составляет более 200. При результате 60-199 возможны легкие фенотипические проявления болезни, риск развития патологии в следующем поколении (если показатель диагностирован у женщины).
Лечение синдрома Мартина-Белл
Методы специфической терапии синдрома в настоящее время отсутствуют. Проводится симптоматическое медикаментозное лечение и психолого-педагогическая коррекция. Усилия врачей и специальных психологов направлены на минимизацию эмоционально-поведенческих отклонений, овладение навыками ходьбы, речи и общения, чтения и письма. Медикаментозная терапия включает прием психостимуляторов, антидепрессантов, ноотропов, противоэпилептических средств и гормональных препаратов (при первичной недостаточности яичников). Обучение пациентов проводится по специальным коррекционно-развивающим программам. Для улучшения социальных навыков используются методы когнитивно-поведенческой терапии, групповые тренинги.
Прогноз и профилактика
Синдром Мартина-Белла не имеет осложнений и не сокращает продолжительность жизни больных, поэтому при своевременной и адекватной медико-психолого-педагогической помощи прогноз достаточно благоприятный: пациенты осваивают навыки общения и самообслуживания, обучаются в специальных школах, иногда овладевают рабочими профессиями. Профилактика основана на медико-генетическом консультировании пар из групп риска и пренатальной диагностике синдрома. Эти меры необходимы женщинам с синдромом преждевременного истощения яичников, семьям, в которых диагностированы премутационные состояния FMR1 или выявлены случаи интеллектуальной недостаточности у мальчиков и мужчин.
Синдром фрагильной Х-хромосомы (ломкой Х-хромосомы, ФХС, FRAX-А, синдром Мартина-Белла) - клиника, диагностика
Данное заболевание является второй после синдрома Дауна причиной задержки умственного развития с коэффициентом IQ менее 50 и наиболее частой причиной семейных случаев умственной отсталости и предпосылкой для развития различных поведенческих проблем, включая аутизм и синдром гиперактивности (Percy et al., 1990; Reiss и Freund, 1990; Hagerman и Hagerman, 2002).
Приблизительно в 35% случаев отмечается задержка умственного развития, обычно легкая, а у 15% пациенток отмечается пограничный уровень интеллекта, сложности в обучении или и то и другое (Kemper et al., 1986). Таким образом, ФХС является важной причиной легкой задержки умственного развития у женщин. Кроме того, приблизительно у 10% женщин с нормальным интеллектом, являющихся носительницами патологической хромосомы, могут отмечаться психиатрические заболевания, в особенности аффективное или шизоидное расстройство (Reiss и Freund, 1990).
Полная мутация гена может стать причиной аутизма, неспособности к обучению, тревожных расстройств и задержки умственного развития. Имеются данные о том, что аутизм также встречается у молодых мужчин, являющихся носителями премутантных аллелей. В ходе одного из недавних исследований было продемонстрировано, что приблизительно у 30% пациентов с ФХС отмечается аутизм; среди пациентов с аутизмом (по сравнению с пациентами только с ФХС) выявляются более низкие когнитивные способности, более выраженные проблемы с речью и отклонения поведения.
У пациентов с синдромом фрагильной Х-хромосомы (ФХС) отмечается нарушение регуляции путей, связанных с метаботрофным рецептором глутамата-5; считается, что данное нарушение метаболизма обусловливает характерный фенотип.
б) Распространенность. Синдром фрагильной Х-хромосомы (ФХС) (полная мутация ДНК) встречается с частотой приблизительно 1:4000 (среди мужчин) и 1:8000 (среди женщин), но учитывая, что исследования ДНК населения в целом не проводились, невозможно сделать вывод о достоверности данных сведений (Hagerman и Hagerman, 2002). Носительство премутаций отмечается с частотой 1:750-1000 мужчин и 1:250-350 женщин. С учетом того, что не у всех носителей фрагильной Х-хромосомы уровень IQ ниже 70, и они (не всегда) могут иметь лишь незначительные проблемы с учебой без гендерных различий, истинная распространенность ФХС (полной мутации и премутации с клинически значимыми отклонениями), вероятно, выше.
У большинства мужчин с полной мутацией отмечаются отклонения от умеренной до тяжелой степени выраженности, в то время как у женщин проявления заболевания имеют менее выраженный характер, тем не менее, около трети пациенток имеют коэффициент IQ менее 70.
Распространенность ФХС среди детей с трудностями в обучении от умеренной до тяжелой степени выраженности и без специфических дисморфических проявлений варьирует от 2% до 10% (Slaney et al., 1995).
в) Диагностика. Диагноз синдрома фрагильной Х-хромосомы (ФХС) можно предположить на основании фенотипа; диагноз легко установить в постпубертатном периоде у мальчиков и затруднительно в препубертатном периоде у мальчиков и у девочек-носительниц, без типичных соматических изменений. Крупные уши и макроорхизм, напротив, часто не связаны с фрагильной Х-хромосомой (Hagerman, 1987). Семейный анамнез задержки умственного развития показателен, но обнаруживается только в трети случаев (Simko et al., 1989).
Исследования ДНК в настоящее время надежны и оправдывают затраты (Rousseau et al., 1991, 1994; Oostra et al., 1993; Wang et al., 1993; Hagerman и Hagerman, 2002a). Экспресс-метод реакции антител, позволяющий выявить ген FMRP в лимфоцитах, был предложен в качестве скрининга для выявления полной мутации (Willemsen et al., 1995).
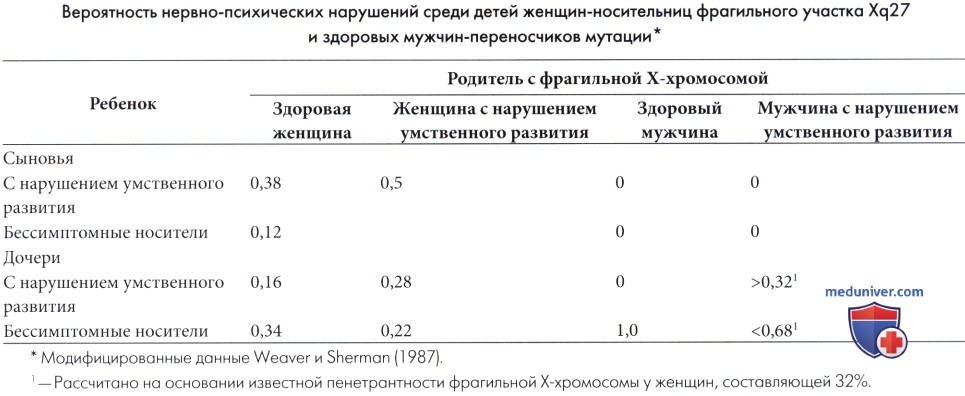
Пренатальная диагностика синдрома фрагильной Х-хромосомы (ФХС) возможна с использованием культуры амниотических клеток или клеток ворсин хориона и методом анализа ДНК. Тем не менее, возникают этические проблемы, так как только у некоторых носителей мутации (особенно среди женщин) в дальнейшем разовьется заболевание. В настоящее время рассчитана (Weaver и Sherman, 1987) вероятность задержки умственного развития у детей больных родителей, но точные рекомендации остаются затруднительными.

Синдром фрагильной Х-хромосомы: удлиненное лицо, большие уши, прогнатизм.
Общее развитие тела адекватное, но отмечается увеличение яичек.
г) Клинические проявления. Заболевание легче выявить у подростков, чем у детей в препубертатном возрасте. Наиболее важными проявлениями являются отсутствие замедленного физического развития, типично связанного с многими причинами умственной отсталости и, в особенности, нормальная или увеличенная окружность головы, вытянутое лицо с выступающей челюстью и макроорхизм (Но et al„ 1989).
В препубертатном возрасте у детей выявляются только следующие признаки: большая голова, большие уши и высокое небо. Помочь в установлении диагноза может также обнаружение повышенной гибкости суставов и мягкие уши. Несмотря на отсутствие макроорхизма у пациентов раннего возраста, в 15-50% случаев отмечается некоторое увеличение яичек (Simko et al., 1989).
Типичные соматические проявления синдрома фрагильной Х-хромосомы (ФХС) представлены в таблице ниже.
Задержка умственного развития при синдроме фрагильной Х-хромосомы (ФХС) обычно выражена слабо или умеренно, но с тенденцией к утяжелению в подростковом и взрослом возрасте (Borghgraef et al., 1987; Hagerman, 1987,1989; Wisniewski et al., 1989). У большинства мужчин по результатам стандартных тестов уровень IQ составляет менее 70 (чаще всего 35-40), но часть пациентов обладает нормальным или низко нормальным коэффициентом IQ. Вербальные навыки обычно превосходят способность к выполнению заданий и зрительно-пространственные навыки.
Эпилепсия является достаточно частым проявлением и встречается в 15-25% случаев (Hagerman и Hagerman, 2002). Приступы часто имеют сложный парциальный характер, относительно доброкачественны и склонны к разрешению в подростковом возрасте, тем не менее, изредка отмечаются тяжелые и сложные случаи эпилепсии. Существует гипотеза о возможной связи припадков с аномалиями червя мозжечка, выявляемыми при ФХС.
В подавляющем большинстве случаев у мужчин отмечаются явные признаки социальной дисфункции. Практически у всех мужчин присутствуют проявления аутизма, но лишь у немногих развивается полный синдром аутизма в сочетании с задержкой умственного развития или без нее. Фрагильная Х-хромосома наиболее распространена среди известных причин аутизма. Часто (приблизительно у половины мальчиков с полной мутацией) встречается гиперкинетический синдром в сочетании с аутизмом или без него, обычно с аутистическими проявлениями (Sullivan et al., 2006). Показатели когнитивных функций при ФХС отличаются от встречающихся при низкофункциональном аутизме, но достаточно часто соответствуют таковым при высокофункциональном аутизме и синдроме Аспергера. Некоторые авторы описывают сочетание синдрома Аспергера и синдрома фрагильной хромосомы (Hagerman, 1989).
В пубертатном периоде часто отмечается остановка когнитивного развития или даже ухудшение когнитивных функций.
Фенотип фрагильной Х-хромосомы также сочетается с синдромом тремора и атаксии (ФХТАС), который представлен тремором, атаксией, периферической поли-нейропатией и когнитивным дефицитом. Обычно отмечается значимая атрофия головного мозга и поражение белого вещества. ФХТАС развивается после подросткового возраста у мужчин (в отдельных случаях у женщин), имеющих премутации. Считается, что данный синдром связан с увеличением уровня аномальной информационной РНК гена FMR1. Имеющаяся в настоящее время информация в сочетании с недавно полученными сведениями о связи выраженности нейропатии (количества внутриядерных включений) и размера премутационного аллеля, подтверждает, что нейродегенеративный фенотип при ФХТАС является следствием распространенности повторов тринуклеотида ЦГГ (Cohen et al., 2006).
Получены убедительные доказательства взаимосвязи премутации гена FMR1 и нарушения функции яичников с потерей фертильности (Wittenberger et al., 2007). Токсическое действие информационной РНК гена FMR1, кодирующей патологический белок, может являться причиной повреждения функции яичников. У женщин с преждевременной недостаточностью яичников отмечается повышенный риск наличия премутации гена FMR1; их информируют о возможности обследования на наличие фрагильной X-хромосомы. Специалисты по репродуктивной медицине могут обеспечить адекватные условия для разъяснения роли обследования на премутацию гена FMR1, создать условия для проведения и своевременно направлять пациенток на генетическую консультацию.
д) Лечение. В настоящее время специфического лечения синдрома фрагильной Х-хромосомы не существует (Hagerman, 1989). Некоторыми врачами использовалась фолиевая кислота (0,5-1,5 мг/кг, дважды в сутки), но эффективность сомнительна. В некоторых отчетах предполагается положительное воздействие фолиевой кислоты на симптомы аутизма, как минимум при назначении в дошкольном возрасте, но после пубертатного периода данный препарат оказывает незначительное отрицательное воздействие или не действует. Многие авторы использовали стимуляторы для подавления гиперактивности (в дозах, рекомендованных детям с синдромом дефицита внимания с гиперактивностью независимо от этиологии) и отмечали благоприятные или хорошие результаты (Hagerman и Hagerman, 2002). Ингибиторы обратного захвата серотонина могут быть эффективны в отношении тревожности, депрессивного настроения и раздражительности, но при применении необходим строгий контроль из-за возможности усиления импульсивности и агрессивного поведения.
е) Другие, связанные с Х-хромосомой синдромы, с задержкой умственного развития. ФХС объясняется только 50% случаев преобладания задержки умственного развития среди мальчиков. Описано множество других более или менее идентифицированных синдромов связанной с Х-хромосомой задержки умственного развития (Opitz et al., 1986). Некоторые пациенты с анэуплоидией половых хромосом имеют трудности в обучении и другие нервно-психические отклонения. Данные заболевания описаны ниже.
1. FRAX-E. В некоторых случаях выявляется второй тип синдрома фрагильной Х-хромосомы (FRAX-E), вызванный распространением повторов тринуклеотида ГЦЦ в области Xq28, 600 тысяч нуклеотидов после гена FMR1 (Flynn et al., 1993). Задержка умственного развития обычно имеет слабо выраженный характер, а во многих случаях полностью отсутствует.
3. Синдром Ренпеннинга. Синдром Ренпеннинга, изначально считавшийся одним из клинических вариантов ФХС, характеризуется задержкой умственного развития от умеренной до тяжелой степени выраженности, легкой микроцефалией, низкорослостью и нормальным строением хромосом (Archidiacono et al, 1987).
4. Синдром Юберга-Марсиди. Данный редкий синдром включает такие проявления как задержка роста, глухота и микрогенитализм (Juberg и Marsidi, 1980).

Актуальность исследования:синдром с ломкой Х-хромосомы особая форма умственной отсталости,сцепленная с полом.Болезнь встречается довольно часто. Особенности фенотипических проявлений:
-Постуберативное увеличение яичек у мужчин
Цель:Согласно популяционным исследованиям,ее распространенность среди белого населения колеблется от 1 на 3717 ло 1 на 8918.Среди больных с умственной отсталостью ломкую хромасому находили у 5.9 % мальчиков и 0.3 %девочек.Ярче всего проявляется у мальчиков.
Задачи : выявить причины болезни,диагностику,этиологию и патогенез.
Краткая история
Еще в начале нашего века клинические исследования выявили значительное преобладание лиц мужского пола среди больных олигофренией. Впервые в 1934 г. J. Martin и J. Bell была описана семья, где умственная отсталость наследовалась по сцепленному с полом типу. В 2-х поколениях большой семьи из Англии насчитывалось 11 умственно отсталых мужчин и 2 женщины с более легким интеллектуальным дефектом. Далее таких семей описывалось все больше, в 1969 г. H. Lubs, проводя цитогенетическое обследование умственно отсталого мальчика из семьи с рецессивным, сцепленным с полом типом наследования, выявил у него вторичную перетяжку на длинном плече Х-хромосомы в области 27-28.
Этиология
Выявленная цитогенетическая аномалия относится к феномену так называемой фрагильности (fragility - ломкость, хрупкость). При данной патологии важно то, что ломкость затрагивает всегда один и тот же участок Х-хромосомы, где локализован мутантный ген. Ни патогенетическая роль выявленного феномена, ни механизм его связи с мутантным геном до сего времени не выяснены. При цитогенетическом исследовании обнаружили не во всех метафазных клетках. Было выявлено, что обнаружение его зависит от определенных условий среды, таких как отсутствие фолиевой кислоты.
Вопрос о наследовании синдрома Мартина-Белл изучен недостаточно, лишь в 1/3 случаев оно характеризуется как типичное Х-сцепленное рецессивного типа. Это означает, что заболевание проявляется исключительно у лиц мужского пола. Генотип мужчины устанавливается по его фенотипу: если он болен, то гемизиготен по аномальному гену, если здоров, то свободен от него. Женщины-носительницы мутантного гена, как правило, фенотипически здоровы. Среди мужчин-родственников матери по женской линии нередко встречаются больные с тем же заболеванием. Среди здоровых членов семьи по женской линии преобладают женщины, также как и среди здоровых сибсов пробанда. Однако в большем числе случаев (2/3) наследование синдрома носит характер нерегулярного сцепленного с Х-хромосомой, а именно получены строгие доказательства возможности передачи маркерной хромосомы здоровыми мужчинами (облигатными носителями), в потомстве дочерей которых отмечаются случаи рождения больных сыновей. Для ген, обуславливающего синдром, определена неполная его пенетрантность у мужчин (79%). Кроме того, женщины-носительницы в ряде случаев (35%) имеют умственную отсталость от легкой до тяжелой ее форм. Хотя матери и дочери мужчин-носителей гена ломкости Х-хромосомы сходны по фенотипу, экспрессия этого гена в их потомстве различна: пенетрантность умственной отсталости выше в потомстве интеллектуально нормальных дочерей мужчин-носителей, чем в потомстве интеллектуально нормальных матерей мужчин-носителей. Эта ситуация, как и передача синдрома здоровыми мужчинами, хорошо объясняется аутосомной супрессорной системой, которая обеспечивает неполную пенетрантность гена.
Ген-супрессор подавляет проявление мутантного гена у облигатного носителя-мужчины с нормальным интеллектом. У его дочерей вследствие рекомбинационных процессов ген-супрессор из системы генотипа исчезает, что является условием для появления мутантного гена у их сыновей. Предположение об аутосомном гене-супрессоре в противоположность супрессору, находящемуся в одной группе сцепления (Х-хромосома) допускает большую частоту их рекомбинаций (независимое комбинирование). Это повышает вероятность такой комбинации генотипа в потомстве, когда мутантный ген входит в систему генотипа без гена-супрессора и формирует соответствующий синдром у носителя.
Существует гипотеза о ломкости Х-хромосомы как о некотором премутационном состоянии локуса Хq27.3, сопровождающимся аномалиями рекомбинации в этом районе. В последующих поколениях они способствуют возникновению мутантного аллеля, обусловливающего синдром Мартина-Белл. Авторы при помощи своей гипотезы объясняют уже упомянутые особенности родословных с нерегулярным наследованием синдрома, согласно которым обеспечивается передача мутантного гена нормальными мужчинами, причем их гетерозиготные дочери не имеют умственной отсталости, у них либо нет ломкого участка, либо количество таких клеток мало. В следующем поколении треть женщин-гетерозигот имеют умственную отсталость и около 29% с ломкой Х-хромосомой. Эти женщины имеют больных сыновей.
Женщины-носительницы мутантного гена могут быть умственно отсталыми, что свидетельствует в пользу того, что мутантный ген не является полностью рецессивным. Пенетрантность умственной отсталости у гетерозигот у равна 30%. Степень умственной отсталости варьирует от легкой до тяжелой, но чаще женщины-носительницы мутантного гена психически нормальны. Изменчивость интеллекта у женщин-носительниц аномального гена ряд авторов объясняют различиями в инактивации ломкой Х-хромосомы в соответствии с гипотезой J. Lejeune, т.е. у женщин с нормальным интеллектом рано реплицирующейся (генетически активной) чаще бывает нормальная Х-хромосома, тогда как у женщин со сниженным интеллектом - ломкая Х-хромосома.
Клиника
Было отмечено, что средняя масса тела пробандов при рождении часто повышена - от 3,5 до 4 кг.
Первым в качестве одного из наиболее выраженных соматических симптомов у подростков и взрослых больных отметили макроорхизм при отсутствии изменений эндокринной функции.
Прежде всего определенные признаки были отмечены в строении лица и тела больных: большая голова с высоким и широким лбом, длинное лицо с увеличенным подбородком, несколько уплощенная средняя часть лица, тупой, слегка клювовидно загнутый кончик носа. Уши большие, иногда оттопыренные, низко расположенные. Кисти и стопы широкие, дистальные фаланги пальцев также широкие, суставы имеют повышенную подвижность. Кожа нередко гиперэластична. Часто встречаются светлоокрашенные радужные оболочки, светлые волосы. Вместе с тем вариабельность фенотипа очень широкая.
Таким образом, выявленные системные соматические изменения затрагивают связочный аппарат, хрящ, кожу, костную систему. Это дает основание предполагать, что в патологический процесс вовлекается соединительная ткань.
По данным практически всех исследователей, неврологическая симптоматика при данном синдроме характеризуется некоторыми слабо выраженными и неспецифическими признаками, которые часто встречаются при умственной отсталости у детей вообще. Чаще всего выявляется мышечная гипотония и дискоординация движений. Кроме того, отмечаются экстрапирамидные (стереотипные гримасы), пирамидные и глазодвигательные нарушения.
Ведущим психопатологическим нарушением является интеллектуальное недоразвитие, которое отмечается всеми исследователями как постоянный синдром. По данным литературы, степень умственной отсталости различна. Вместе с тем есть работы, где описываются больные, имеющие пограничную умственную отсталость и даже с интеллектом, соответствующим норме.
Было обращено внимание на своеобразную речь этих пациентов. Позднее речь пациентов стали называть "повторяющейся, персеверативной, бормочущей". К ее особенностям относится ускоренный темп и выраженные эхолалии и персеверации. Она является весьма характерной именно для этой патологии и может иметь определенное диагностическое значение.
Часто выявляются разнообразные нарушения поведения: синдром двигательной расторможенности, выраженная аффективная возбудимость, агрессивность.
В качестве одной из частых психопатологических особенностей отмечена шизофреноподобная симптоматика, включающая в себя подпрыгивания, похлопывания руками, повороты вокруг своей оси, встряхивание кистями, "манежный" бег, разнообразные гримасы, монотонное хныканье.
Помимо умственной отсталости, чаще всего у больных отмечаются проявления раннего детского аутизма (отсутствие потребности в контактах с окружающими, отгороженность от внешнего мира, слабость эмоционального реагирования по отношению к близким , недостаточность реакций на зрительные и слуховые раздражители, приверженность к сохранению неизменности окружающего, боязнь всего нового, однообразность поведения со склонностью к стереотипным примитивным движениям, разнообразные расстройства речи, непереносимость взгляда в глаза, взгляд "мимо" и "сквозь" людей.
Методы диагностики
Клинический. К минимальным диагностическим признакам относятся: умеренная или глубокая умственная отсталость (97,5%), большие оттопыренные ушные раковины (62%), выступающий лоб и массивный подбородок, макроорхизм (68,4%), характерная речь (41,4%).
Цитогенетический. Существует стандартный метод изучения культуры лимфоцитов периферической крови больных. Культуру получают на среде 199 (дефицитной по фолиевой кислоте) с добавлением 15% сыворотки крупного рогатого скота. Далее через 72 часа ее фиксируют и окрашивают с помощью трипсин-гимза. Анализируют на стадии метафазы, учитывают число клеток с ломкой Х-хромосомой на 100 изученных клеток.
Были описаны характерные для синдрома изменения на ЭЭГ: отсутствие a-ритма, значительное усиление q-диапазона, преобладание нерегулярной q- и D-активности в затылочных зонах коры, в центральных, лобных и теменных отделах полушарий отмечено доминирование высокоамплитудного q-ритма.
При анализе дерматоглифических данных с помощью разработанной дискриминантной классификационной функции их диагностическая значимость составляет 80%.
Подходы к лечению
В связи с тем, что ломкость Х-хромосомы можно выявить лишь в среде, обедненной фолатами, возникло предположение о роли дефицита фолиевой кислоты в патогенезе самого заболевания. Это привело к попыткам лечить умственную отсталость введение фолатов.
Эффект от лечения был более выражен у детей, чем у взрослых. В целом все авторы отмечают, что у больных при лечении препаратами фолиевой кислоты не происходит улучшения в интеллектуальном развитии, но улучшается поведение: уменьшаются гиперактивность, агрессивность, повышается внимание, моторная координация, улучшается качественная и количественная сторона речи.
Было предложено применять психостимуляторы. Авторы отмечают неплохие результаты, особенно при лечении метилфенидатом. Дети становились более внимательными, сосредоточенными, у них уменьшалась двигательная активность.
В 1981 г. ломкую Х-хромосому впервые обнаружили в культуре амниотических клеток. Более точные результаты получаются при исследовании крови плода, чем при биопсии плаценты.
Профилактика
Рождения больного ребенка существенно различается в зависимости от того, унаследована ли мутация или же она возникла de novo. В последнем случае риск рождения больного ребенка крайне мал. "Свежесть" мутации можно предположить в том случае, если среди родственников нет больных мужчин и не отмечены случаи умственной отсталости у женщин, при этом у родственников тест на ломкую Х-хромосому отрицателен. Но, учитывая неполную пенетрантность гена, точно доказать спорадический случай невозможно, поэтому несмотря на единичный случай заболевания в семье, не следует утверждать, что мутация возникла de novo. Исходя из этого ребенка считают сегрегантом, а мать - гетерозиготой по аномальному гену, и следующую беременность рекомендуют вести под контролем ломкости Х-хромосомы у плода.
Лечение
Лечения для синдрома ломкой Х-хромосомы не существует, однако есть надежда, что дальнейшие исследования причин заболевания предоставят новые возможности терапии. В настоящее время симптомы можно облегчить с помощью когнитивно-поведенческой терапии, специфического обучения, медикаментов и, при необходимости, лечения физических аномалий. Лица, имеющие случаи синдрома ломкой Х-хромосомы в семье, должны получить генетическое консультирование при планировании беременности
Поскольку в эксперименте обнаружение ломкости удалось обнаружить в среде, бедной фолатами, было предложено лечить таких детей фолиевой кислотой.
Эффект от лечения у детей выражен больше, чем у взрослых: пропадает агрессия, повышается внимание, улучшается моторика и речь.
Также пробуют лечить таких больных психостимуляторами.
GeneTest.Информационный ресурс по медицинской генетикех Университет Вашинктона.Сиэттл 1993-2007
OMIM:Менделирующее наследование у человека online.Институн генетической медицины McKusick-Nathans Университет Джона Хопкинса.Национальный центр биотехнологической информации.Национальная медицинская библиотека
Роберт Л.Ньюссбаум,Родерик Р. Макс-Иннес, Хантингтон Ф.Виллард Медицинская генетика 2010
Кеннет Л.Джонс Наследственные синдромы по Дэвиду Смиту Атлас-справочник.Москва 2011
Читайте также: