
Мутационное заболевание короткий доклад
Обновлено: 01.05.2024
Виды мутаций у человека. Варианты
Последовательность ядерной ДНК у любых двух человек идентична почти на 99,9%. Только очень небольшая доля последовательности ДНК различается у разных людей, обеспечивая генетическую изменчивость. Некоторые различия в последовательности ДНК не имеют влияния на фенотип, тогда как другие — непосредственные причины болезней. Между двумя крайностями — изменения, ответственные за генетически предопределенную фенотипическую изменчивость в анатомии и физиологии, переносимость пищи, реакции на лечение или побочные эффекты медикаментов, восприимчивость к инфекциям, склонность к опухолям и, возможно, даже изменчивость в различных чертах личности, спортивных способностях и художественном таланте.
Одно из важных понятий генетики человека и медицинской генетики — то, что генетические болезни — только наиболее очевидное и часто крайнее проявление генетических различий, один конец непрерывного спектра изменений от редких вариантов, вызывающих болезнь, через более частые варианты, увеличивающие восприимчивость к болезни, до наиболее частых изменений, не имеющих явного отношения к болезни.
Виды мутаций у человека
Мутация — любое изменение в последовательности нуклеотидов или расположения ДНК. Мутации можно классифицировать на три категории: влияющие на количество хромосом в клетке (геномные мутации), изменяющие структуру отдельных хромосом (хромосомные мутации) и изменяющие индивидуальные гены (генные мутации). Геномные мутации — изменения числа неповрежденных хромосом (анеуплоидии), возникающие вследствие ошибок в расхождении хромосом в мейозе или митозе.

Хромосомные мутации — изменения, затрагивающие только часть хромосомы, например частичные дупликации, делеции, инверсии и транслокации, которые могут происходить спонтанно или возникать вследствие аномального расхождения транслоцированных хромосом в ходе мейоза. Генные мутации — изменения в последовательности ДНК ядерного или митохондриального генома, от мутации в единственном нуклеотиде до изменений, захватывающих много миллионов пар оснований. Множество типов мутаций представлены разнообразными аллелями в отдельных локусах при более чем тысяче разных генетических заболеваний, а также среди миллионов вариантов ДНК, обнаруживаемых во всем геноме в нормальной популяции.
Описание разных мутаций не только увеличивает осведомленность о генетическом разнообразии человека и хрупкости человеческого генетического наследия, но также содействует получению информации, необходимой для обнаружения и скрининга генетических болезней в конкретных семьях риска, а также — для некоторых болезней — в популяции в целом.
Геномная мутация, приводящая к утрате или дублированию целой хромосомы, изменяет дозу и, таким образом, уровень экспрессии сотен или тысяч генов. Аналогично затрагивающая большую часть одной или нескольких хромосом хромосомная мутация также может влиять на экспрессию сотен генов. Даже небольшая генная мутация может иметь большие последствия, в зависимости от того, какой ген затронут и к чему приводит изменение в экспрессии этого гена. Мутация гена в виде изменения единственного нуклеотида в кодирующей последовательности может вести к полной утере экспрессии гена или образованию белка с измененными свойствами.
Некоторые изменения ДНК, тем не менее, не имеют фенотипических эффектов. Хромосомная транслокация или инверсия может не влиять на критическую часть генома и абсолютно не иметь фенотипических эффектов. Мутация в пределах гена может не иметь эффекта вследствие того, что либо не изменяет аминокислотную последовательность полипептида, либо, даже если это происходит, изменение в закодированной аминокислотной последовательности не изменяет функциональные свойства белка. Следовательно, не все мутации имеют клинические последствия.
Все три типа мутаций происходят со значимой частотой во множестве разных клеток. Если мутация происходит в ДНК половых клеток, она может передаваться последующим поколениям. В отличие от этого, соматические мутации происходят случайным образом только в части клеток определенных тканей, приводя к соматическому мозаицизму, наблюдаемому, например, при многих опухолях. Соматические мутации не могут передаваться последующим поколениям.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021

Наследственные заболевания передаются от одного или обоих родителей детям. Они вызываются генетическими мутациями, но далеко не все генетические заболевания являются наследственными. Как в этом разобраться, какие виды заболеваний бывают, как их лечить и как диагностировать — рассказываем в нашей статье.
Содержание
Что такое наследственные заболевания?
Наследственные заболевания — это заболевания, обусловленные генными или хромосомными мутациями. У людей от 20 000 до 25 000 генов. Генетическая мутация возникает, когда изменяется один или несколько генов. Если это генетическое изменение передается детям, то это наследственное генетическое заболевание.
При совпадении у партнеров статусов носительства определенных болезней есть высокий риск рождения ребенка с наследственным заболеванием. Если у вас не проявляются симптомы заболевания, вы по-прежнему можете быть носителем и передать мутации своим детям.
Многие генетически обусловленные заболевания проявляются не сразу после рождения, а спустя некоторое время. От наследственных заболеваний следует отличать врожденные заболевания, вызванные внутриутробными повреждениями, например, инфекцией или внешними воздействиями.
Чем отличаются наследственные заболевания от врожденных нарушений?
Генетические заболевания являются результатом изменения одного или нескольких генов и могут передаваться в поколениях или нет.
Все наследственные заболевания имеют генетическое происхождение, т. е. являются результатом изменения одного или нескольких генов и передаются из поколения в поколение. Симптомы могут не проявляться с самого рождения.
Врожденные нарушения могут быть наследственными или нет, а симптомы могут проявляться с рождения. Но их появление не обязательно связано с генетикой.
Виды наследственных заболеваний
Наследственные заболевания разделяются на хромосомные, генные и митохондриальные.
Хромосомные заболевания
В настоящее время описано около 1000 форм хромосомных заболеваний. Хромосомные заболевания возникают в результате изменения числа или структуры хромосом. Они характеризуются общими признаками: маленькая масса и длина тела при рождении, отставание в умственном и физическом развитии, задержка и аномалии полового развития и прочее.
Моногенные заболевания
Моногенные заболевания возникают в результате повреждения ДНК на уровне гена. Количество моногенных заболеваний по некоторым оценкам достигает 5000.
Среди признаков моногенных болезней можно выделить: различные формы умственной отсталости, дефекты органов слуха, зрения, скелетные дисплазии, болезни нервной, эндокринной, иммунной и других систем. К числу наиболее известных моногенных болезней относятся муковисцидоз, гемофилия А и В, болезнь Гоше, миодистрофия Дюшенна/Беккера, спинальная мышечная атрофия, дальтонизм.
Выявить тяжелые моногенные заболевания можно с помощью пренатальной диагностики, а также, определив наличие мутаций у родителей с помощью генетического теста.
Интереснее всего мне было узнать об особенностях метаболизма. Именно поэтому я выбрала Атлас: только тут есть достаточно объемный раздел на эту тему. Например, всю жизнь я борюсь с весом, мигренью, болями в шее и спине, анемией.
Митохондриальные заболевания
Митохондриальные заболевания обусловлены генетическими, структурными, биохимическими дефектами в функционировании митохондрий, которые приводят к нарушению тканевого дыхания.
Митохондрии содержат свою собственную ДНК. А болезни, вызванные мутациями в митохондриальной ДНК, наследуются исключительно по материнской линии. Если именно таким образом было унаследовано митохондриальное заболевание, существует 100% вероятность того, что каждый ребенок в семье его унаследует.
Симптомы могут включать в себя: нарушение роста, слабость мышц, аутизм, ментальные расстройства, проблемы с дыханием, слухом и зрением. Примеры митохондриальных заболеваний: синдром Лея, синдром Вольфа-Паркинсона-Уайта, наследственная оптическая нейропатия Лебера и другие.
Полигенные или мультифакториальные заболевания
Существуют также болезни с наследственной предрасположенностью, которые называют мультифакториальными или полигенными заболеваниями.
Мультифакториальные заболевания обусловлены наследственными факторами риска, и в значительной степени — неблагоприятным воздействием среды. К мультифакториальным заболеваниям относятся большинство хронических заболеваний, включая сердечно-сосудистые, эндокринные, иммунные, нервно-психические, онкологические и др. Например, бронхиальная астма, сахарный диабет, ревматоидный артрит, гипертоническая болезнь сердца и т.д.
Как передаются наследственные заболевания?
Организм человека состоит из триллионов клеток. Каждая клетка имеет ядро, которое содержит хромосомы. Каждая хромосома состоит из плотно свернутых нитей дезоксирибонуклеиновой кислоты (ДНК).
Гены — это инструкции по сборке белков в нашем организме, которые определяют специфические черты каждого человека, например, цвет глаз или волос. Большинство клеток в организме обычно содержат 46 хромосом, организованных в 23 пары. В каждой из этих 23 пар есть одна унаследованная хромосома от отца и одна — от матери. Из 23 пар 22 пары одинаковые у женских и мужских организмов, а одна оставшаяся определяет, являетесь вы мужчиной (XY) или женщиной (XX).
Мутации, из-за которых возникают наследственные заболевания, могут иметь доминантный или рецессивный характер наследования.
Доминантное наследование означает, что только одна копия гена — от матери или отца — должна иметь мутацию (или патогенный вариант гена) для проявления признака или заболевания. А при рецессивном типе человек наследует две измененные копии одного и того же гена.
Аутосомно-доминантный паттерн наследования
При аутосомно-доминантном наследовании заболеваний генетически обусловленная болезнь проявляется в том случае, если у человека есть хотя бы один мутированный ген, и этот ген не расположен на половых (Х и Y) хромосомах.
Болезнь Хантингтона и синдром Марфана — два примера аутосомно-доминантных болезней. Мутации в генах BRCA1 и BRCA2, которые также связаны с раком молочной железы, передаются по этой схеме.
Аутосомно-рецессивный паттерн наследования
При аутосомно-рецессивном наследовании мутируют обе копии генов. Чтобы унаследовать аутосомно — рецессивное заболевание, такое как муковисцидоз, спинальная мышечная атрофия, или фенилкетонурия (ФКУ), оба родителя должны быть носителями. Ребенок наследует две копии дефектного гена — по одной от каждого родителя. Например, люди, имеющие одну копию гена с мутацией, а вторую — без мутации, называются носителями, потому что сами они здоровы.
Х-сцепленное рецессивное наследование
В Х-сцепленном рецессивном наследовании мутированный ген находится на Х-хромосоме. Болезнь проявляется только в случае, если другой Х-хромосомы с нормальной копией того же гена у человека нет.
Мышечная дистрофия Дюшенна, некоторые виды дальтонизма и гемофилия А — примеры рецессивных заболеваний, связанных с X-хромосомой. Мужчина с рецессивным заболеванием, связанным с X-хромосомой, передаст свою нетронутую Y-хромосому сыновьям, и ни один из них не пострадает. Если он передаст свою Х-хромосому (с дефектным геном) своим дочерям, то все они будут носителями болезни. У его дочерей может не быть симптомов или только легкие признаки заболевания, но они могут передать мутированный ген своим детям.
Женщины-носители рецессивного заболевания, связанного с X-хромосомой, часто имеют лёгкие признаки заболевания или вообще не имеют симптомов. Это связано с тем, что у женщин-носителей есть одна нормальная копия гена и одна мутированная копия. Нормальная копия обычно компенсирует дефектную копию в женском организме, в отличие от мужчин, у которых только одна X-хромосома.
Женщины, имеющие только один патологический ген, передают заболевание в среднем половине своих детей вне зависимости от пола. Женщины же, имеющие два патологических гена, передают заболевание всем своим детям. К таким заболеваниям относятся гемофилия А и дальтонизм.

Как генетическое тестирование помогает при планировании семьи
Если вы знаете или предполагаете, что у вас или вашего партнера в семейной истории есть какое-либо генетическое заболевание, вы можете определить это с помощью Генетического теста Атлас. Генетическое консультирование поможет вам узнать о методах лечения, профилактических мерах и репродуктивных возможностях.
Как лечить наследственные заболевания и как с ними жить?
Раньше наследственные заболевания были неизлечимы. Сейчас это по-прежнему остаётся проблемой для многих заболеваний, но для некоторых из них методы лечения уже найдены. Например, это касается болезней, связанных с нарушением метаболизма.
При большинстве наследственных нарушений обмена веществ один фермент либо вообще не вырабатывается организмом, либо вырабатывается в форме, которая не работает. Например, при отсутствии какого-либо фермента в организме могут накапливаться токсичные вещества или может не синтезироваться необходимый продукт — как при гемохроматозе 1 типа.
При этом заболевании организм поглощает слишком много железа из пищи и не может естественным образом избавиться от избытка. Это может привести к чрезмерному накоплению железа в сердце, поджелудочной железе и печени.
Лечение генетических нарушений обмена веществ следует двум общим принципам:
- Необходимо сократить или исключить прием любой пищи или лекарств, которые не усваиваются организмом.
- Заменить или восполнить отсутствующий или неактивный фермент для восстановления метаболизма с помощью диеты и/или лекарств.
Есть более серьезные и распространенные наследственные заболевания, которые не лечатся. Например, мековисцидоз — скопление слизи в лёгких и в пищеварительной системе. От муковисцидоза нет лекарства, но разные методы контроля симптомов помогают предотвращать или уменьшать осложнения и облегчать жизнь с этим заболеванием.
Со временем муковисцидоз прогрессирует и может привести к летальному исходу, особенно при наличии сопутствующих инфекций. Сегодня благодаря достижениям медицины около половины людей с муковисцидозом доживают до 40 лет. Дети, рожденные с этим заболеванием в наши дни, смогут прожить ещё дольше.
Одно из самых тяжелых наследственных заболеваний, спинальная мышечная атрофия, также с недавнего времени поддается лечению с помощью генной терапии. Но доступен этот метод далеко не каждому. Препарат для лечения СМА — самый дорогой лекарственный препарат в мире.
Как я могу узнать, что являюсь носителем генетического заболевания?
Наши гены содержат инструкции, которые сообщают организму, как правильно функционировать. При изменении этих инструкций развиваются различные заболевания. Во многих случаях симптомы впервые проявляются в зрелом возрасте, поэтому иногда мы не знаем, что являемся носителями. Предупредить риски развития и передачи наследственного заболевания можно с помощью Генетического теста Атлас.

Заболевания, вызванные генными и хромосомными мутациями, называются наследственными. Не следует путать их с врожденными заболеваниями: наследственные заболевания обусловлены внутренними причинами, а потому они отличаются от врожденных тем, что последние возникают из-за внешнего воздействия, которое повреждает эмбрион. Пример такого воздействия – инфекция у матери. Также врожденные заболевания проявляются уже при рождении, а наследственные могут проявить себя спустя несколько лет, даже во взрослом возрасте.
Также наследственные болезни отличаются от того, что называется наследственной предрасположенностью. Такая предрасположенность возникает, если в роду у человека есть кровные родственники, страдающие такими заболеваниями, как сахарный диабет, язва желудка, атеросклероз, рак, ожирение и так далее. Наследственная предрасположенность может возникнуть под влиянием некоторых факторов, а может и не проявить себя никак.
Но наследственные заболевания – это передача генетической или хромосомной особенности работы организма.
Что такое генные болезни
Генные болезни наиболее распространены в категории наследственных. Например, ферментопатия – это такое изменение свойств ферментов в результате генной мутации, когда фермент либо вообще не вырабатывается организмом, либо сильно изменяет свои свойства. Из-за этого в организме не могут происходит определенные биологические реакции. Нарушение жирового обмена, обмена пуринов, углеводов, металлов, аминокислот – пример таких генных болезней.
Примеры генных болезней
Фенилкетонурия: заболевание, передающееся из поколения в поколение. Организмом не усваивается аминокислота фенилаланин, отвечающая за выработку адреналина, норадреналина и тирозина. В результате поражается нервная система, нарушаются двигательные функции и наступает слабоумие.
Синдром Марфана (арахнодактилия): поражение соединительной ткани в результате мутации гена. Костно-мышечная система, глаза, кожа, сердечно-сосудистая система сильно поражены. Высокий рост, сильная худоба, длинные руки и ноги, чрезмерная подвижность суставов, деформация грудной клетки и позвоночника – все это делает человека похожим на паука. Корней Чуковский и Авраам Линкольн имели это наследственное заболевание.
Болезнь Альцгеймера: генная мутация, хорошо известная всем, развивается ближе к 50-ти годам.
Примеры хромосомных болезней
Изменения числа и строения хромосом при формировании половых клеток называется хромосомными мутациями. Эти мутации приводят часто к выкидышу или ребенок рождается мертвым.
Синдром Дауна: лишняя хромосома в хромосомном наборе приводит к болезни, выражающейся в умственной отсталости, низкой сопротивляемости организма и своеобразном внешнем виде.
Синдром Шерешевского–Тернера: отсутствие одной половой хромосомы. Болезнь поражает только женщин, у которых яичники оказываются недоразвитыми, а внешние половые признаки сглажены. При этом женщина эмоционально неустойчива, но умственное развитие в норме, менструации отсутствуют, нет возможности иметь детей.
Синдром Клайнфельтера: наличие у мужчины одной или больше женских половых хромосом. Мужчина обладает женственной внешностью, яички недоразвиты и сперматозоиды не образуются.

Наследственные заболевания — заболевания, этиологическим фактором которого является мутация. Хромосомные болезни — группа наследственных заболеваний, обусловленных изменением числа и структуры хромосом.
Частота хромосомных болезней составляет — 5-7 на 1000 новорожденных.
Различают следующие хромосомные нарушения:
1). Болезни, связанные с нарушением числа аутосом (анеуплоидии, триплоидии и др.).
2). Болезни, связанные с изменением числа хромосом (трисомиии, моносомии).
3). Болезни, связанные с изменением структуры хромосом (делеции, дупликации, инверсии, транс локации: сбалансированные и несбалансированные).
Наследственные заболевания, обусловленные микроделециями и микродупликациями хромосом (болезни геномного импритинга).
Моногенные болезни — заболевания, обусловленные мутациями на уровне гена.
Тип наследования — аутосомно доминантный, аутосомно-рецессивный, Х-сцепленный доминантный, Х-сцепленный рецессивный.
Митохондриальные заболевания — передаются от матери с митохондриальной ДНК.
Мультифакториальные заболевания, врожденные пороки развития.
В мире описано более 300 наследственных заболеваний с нарушением слуха, различными формами тугоухости, пороками развития ушной раковины.
В статье описаны наиболее часто встречающиеся заболевания, их основные клинические проявления.
Синдром Гольденхара (гемифациальная микросомия, окулоаурикуловертебральная дисплазия) Omim 164210.
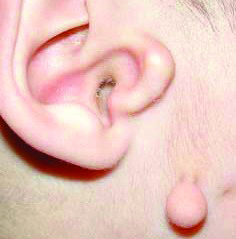
Частота развития заболевания — 1:3000 — 1:5000 новорожденных. Этиология заболевания остается до конца неизвестной. Большинство случаев — спорадические. Хотя выявлены семьи, у которых этот синдром передается по аутосомно-рецессивному и аутосомно-доминантному типу наследования. У нескольких детей, имеющих клиническую симптоматику, сходную с Синдромом Гольденхара, были выявлены хромосомные перестройки на 5, 8 и 21 хромосомах. В нескольких спорадических случаях синдрома Гольденхара были выявлены мутации в Гене GSC, который является кандидатным геном для гена HFM. У пяти пациентов — в гене TCOF1, мутации в котором приводят к развитию синдрома Тричера-Коллинза. Основные клинические проявления заболевания: Гипоплазия скуловой или нижниечелюстной области; Макростомия, асимметрия рта (вытянут в оду сторону); Гипоплазия мимических мышц; Микротия; Преаурикулярные выросты, ямки; Атрезия наружного слухового прохода; Кондуктивная тугоухость; Нейросенсорная тугоухость; Эпибульбарные дермоиды; Колобомы верхнего века; Микрофтальм; В 70% случаев поражение одностороннее; Гипоплазия позвонков; Клиновидные позвонки; В некоторых случаях — умственная отсталость, врожденные пороки сердца.
Гемифациальная микросомия с пороками конечностей (141400 Omim).
Тип наследования предположительно аутосомно-доминантный; Большинство случаев — спорадические; Врожденные пороки конечностей; Трехфаланговые пальцы; Удвоение фаланг; Расщелина мягкого и твердого неба; Гемифациальная микросомия; Преаурикулярные выросты; Кожные выросты в области нижней челюсти; Микротия; Атрезия наружного слухового прохода; Кондуктивная тугоухость; Данный синдром может рассматриваться как вариант Синдрома Гольденхара.
Бранхио-ото-ренальный синдром (Синдром Менльника-Фрейзера)(Omim 113650).
Тип наследования — аутосомно-доминантный; Заболевание обусловлено мутациями в гене EYA1, расположенного на длинном плече хромосомы 8 (8q13.3); Частота заболевания 1:40000 новорожденных; Длинное лицо; У 10 % пациентов -парез лицевого нерва; Нейросенсорная тугоухость (20%); Кондуктивная тугоухость (30%); Смешанная тугоухость (50%); Микротия; Чашеобразные уши; Деформация мочки, наружного уха; Бранхиогенные кисты или свищи; Стеной наружного слухового прохода; Пороки развития среднего и внутреннего уха; Атрезия или сужение слезного канала; Дисплазия Мондини; Прогнатия; Высокое небо; Расщелины твердого и мягкого неба; Расщепление небного язычка; Жаберные щели, расположенные симметрично с обеих сторон — редко; Дисплазия почек; Аплазия почек; Поликистоз почек; Пороки собирательной системы почек; Врожденный вывих бедра; Незавершенный поворот кишечника.
Бранхио-отический синдром (Omim 602588).
Тип наследования — аутосомно-доминантный; Заболевание обусловлено мутациями в гене EYA1; Нейросенсорная тугоухость; Кондуктивная тугоухость; Смешанная тугоухость; Микротия; Чашеобразные уши; Деформация мочки, наружного уха; Бранхиогенные кисты или свищи; Стеной наружного слухового прохода; Пороки развития среднего и внутреннего уха; Данный синдром иногда рассматривают, как вариант синдрома Мельника -Фрезера, но большинство исследователей склоняются к тому, что это разные заболевания, вызванные аллельными мутациями гена EYA1.
Бранхио-окуло-фациальный синдром (Omim 113620).
Тип наследования — аутосомно-доминантный. Заболевание обусловлено мутациями в гене TFAP2A, расположенного на коротком плече 6 хромосомы — 6р24.3. Основные клинические проявления заболевания: Пренатальная задержка роста; Задержка роста; Бронхиогенные кисты или свищи; Атрофия или очаговая аплазия кожи; Гемангиомы на коже; Атрезия слезного протока; Колобомы; Микрофтальм; Анофтальмия; Гамартома сетчатки; Дермоидные кисты глазницы; Эпителиальные кисты радужной оболочки; Катаркта; Миопия; Монголоидный разрез глаз; Телекант; Миопия; Низкопосаженные, ротированнын ушные раковины; Гипоплазия верхней части завитка; Микротия; Преаурикулярные выросты; Кондуктивная тугоухость; Аномалии среднего уха; Свищи над ушами; Широкое, запавшее переносье; Псвевдо-расщелины, расщелины губ, неба; Аномалии зубов; Микрогнатия; Гипоплазия скуловых костей; Агенезия почек; Дисплазия почек; Клинодактилия, полидактилия, грубое нарушение дерматоглифики; Гипоплазия ногтей; Эктопия тимуса; Ранняя седина; Умственная отсталость легкой степни; Гнусавый голос.
Синдром микротии нарушения слуха и расщелины неба (Omim 612290).
Тип наследования: аутосомно-доминантный, аутосомно-рецессивный. Заболевание обусловлено мутациями в гене HOXA2, расположенном на коротком плече 7 хромосомы — 7р15.2. Основные клинические проявления заболевания: Микротия; Сужение наружного слухового прохода (выраженное — при аутосомно-рецессивных формах); Деформация слуховых косточек; Заполнение барабанной полости костной тканью; Сращение слуховых косточек (при аутосомно-рецессивных формах); Кондуктивная тугоухость; Полная потеря слуха (при аутосомно-рецессивных формах); Расщелина твердого и мягкого неба.
Синдром Таунза-Брукса (Omim 107408).
Тип наследования — аутосомно-доминантный. Заболевание обусловлено мутациями в гене SALL1, который расположен на длинном плече 16 хромосомы 16q12.1. Основные клинические проявления заболевания: Нейросенсорная тугоухость; Смешенная тугоухость; Маленькие, чашеобразные уши со свисающим завитком; Преаурикулярные выросты; Трехфаланговый, раздвоеннный широкий 1 палец; Преаксиальная полидактилия; Псевдоэпифиз 2-ой пястной кост; Сращение костей кистей и стоп; Гипоплазия или аплазия 3 пальцев; Клинодактилия 5 пальцев; Эктопия или атрезия заднего прохода; Врожденные пороки влагалища и прямой кишки; Агенезия или гипоплазия почек, поликистоз почек; Пузырно-мочеточниковый рефлюкс; Расщепление мошонки; Гипоспадия; Иногда — умственная отсталость.
Синдром Фрейзера (синдром Фрайзера) (Omim 219000).
Тип наследования — аутосомно-рецессивный. Заболевание обусловлено мутациями в генах FRAS1, расположенного на длинном плече 4 хромосомы — 4q21.21, GRIP1, расположенного на длинном плече 12 хромосомы — 12q14.3, и гена FREM2, расположенного на длинном плече 13 хромосомы — 13q13.3. Основные клинические проявления заболевания: Двусторонний криптофтальм; Недоразвитие глазного яблока; Низкая граница роста волос на лбу на стороне поражения; Западение лобной кости на стороне поражения; Широкий нос; Запавшее переносье; Недоразвитие крыльев носа; Чашеобразные ушные раковины; Микроотия; Атрезия слухового прохода; Пороки среднего уха; Кондуктивная тугоухость; Расщелина твердого и мягкого неба; Стеноз трахеи; Атрезия трахеи; Кожная синдактилия на кистях и стопах; Крипоизм; Гипоспадия; Атрезия влагалища; Агенезия почек; Микроцефалия; Менингоцеле; Энцефалоцеле; Более 20% детей погибают в первый год жизни.
Синдром Тричера-Коллинза (Синдром Франческетти, челюстно-лицевой дизостоз) (Omim 154500).
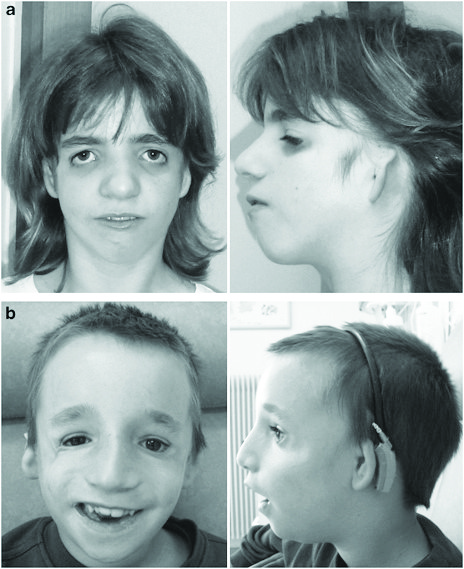
Тип наследования — аутосомно-доминантный. Заболевание обусловлено мутациями в гене TCQF1, расположенного на длинном плече 5 хромосомы — 5q31.3-q32. В настоящее время три типа данного заболевания. Основные клинические проявления заболевания: Антимонголоидный разрез глаз; Гипоплазия скуловых костей; Микрогнатия; Колобома нижнего века; Отсутствие ресниц на нижнем веке; Аномалии ушных раковин; ВПР наружного слухового прохода; Кондуктивная тугоухость; Расщелина неба; Нарушения глотания; Низкая граница роста волос на висках; Гипоплазия средней трети лица; Врожденные пороки сердца; Крипторхизм.
Синдром Пендреда (Omim 274600).
Тип наследования — аутосомно-рецессивный. Заболевание обусловлено мутациями в гене SLC26A4, расположенном на длинном плече 7 хромосомы, 7 q22.3. Основные клинические проявления заболевания: Нейросенсорная тугоухость; Аномалии развития структур костного лабиринта внутреннего уха; Неполное разделение завитка улитки; Дисплазии лабиринта улитки; Широкий водопровод преддверия; В подростковом возрасте — увеличение размеров щитовидной железы; Гипотиреоз; Карцинома щитовидной железы.
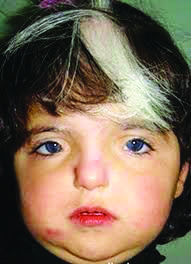
Тип наследования для I, II и III типов — аутосомно-доминантный, для четвертого — аутосомно-рецессивный. Основные клинические проявления заболевания: Короткие глазные щели; Телекант; Нос прямой; Переносье широкое, высокое; Гипоплазированные крылья носа; Густые брови; Синофриз; Очаги депигментации на коже; Бесцветные ресницы и брови; На глазном дне — картина, характерная для альбинизма; Седая прядь волос на лбу; Аплазия заднего полукружного канала; Нейросенсорная тугоухость; Выступающая нижняя челюсть; Высокое стояние лопаток; Добавочные ребра; Spina bifida; Врожденные пороки сердца.
В заключение хотелось бы сказать, что при подозрении у ребенка на наследственную патологию, сопровождающуюся различными формами нарушения слуха, ребенок должен получить консультацию врача-генетика, и при необходимости ему будет проведено молекулярно-генетическое обследование, исследование кариотипа для установления точного диагноза наследственного заболевания, уточнения прогноза развития заболевания и возможности медико-генетического консультирования семьи.
Читайте также: