
Атаксия фридрейха генетика доклад
Обновлено: 02.07.2024
Симптомы атаксии Фридрейха чаще появляются на первом, втором десятилетии жизни, изредка на третьем и четвертом десятилетии. Появляется неуверенность, пошатывание, спотыкание при ходьбе, частые падения, нарушается почерк из-за тремора, появляется дизартрия, слабость в ногах, нарушается слух. Исчезают сухожильные и надкостничные рефлексы (в первую очередь ахиловые и коленные). Иногда ранним симптомом может быть ревмокардит. Больные не выполняют пяточно-коленную пробу, появляется покачивание в позе Ромберга, которое усиливается при закрывании глаз, расстройства сидения. Симптом Бабинского. Часто нистагм.
Постепенно нарушается глубокая чувствительность, нарастает мышечная атрофия, на начальных этапах более выражена на нижних конечностях, с течением болезни захватывает и верхние. Формируется тотальная арефлексия. Атрофируется зрительный нерв, развивается катаракта, что ведет к слепоте, нарушается функция тазовых органов, развивается деменция.
Течение болезни неуклонно прогрессирующее, при отсутствии адекватного лечения, длительность болезни обычно не превышает 20 лет. Непосредственной причиной смерти могут быть сердечная и легочная недостаточность, инфекционные осложнения. В редких случаях при отсутствии сахарного диабета и сердечных нарушений, больные доживают до 70-80 лет. Прогноз благоприятнее у женщин: более 20 лет с начала заболевания живут 100% женщин и только 63% мужчин.
Диагностика: Компьютерная томография головного мозга, которая остается основной диагностикой атаксий при этом заболевании малоэффективна, т.к. обнаруживает изменения только на поздних стадиях. Удается обнаружить только слабую степень атрофии мозжечка на ранней стадии и атрофию полушарий, расширение стволовых цистерн, боковых желудочков и субарахноидального пространства обоих полушарий на более поздних стадиях. Ранняя диагностика атаксии Фридрейха производится с помощью МР-томографии, которая дает возможность обнаружить атрофию спинного мозга и уменьшение поперечного размера спинного мозга, особенно усиливающееся в каудальном направлении на развернутой стадии, и умерено выраженную атрофию моста, мозжечка и продолговатого мозга. На начальной стадии обязательно проводится электрофизиологическое исследование, при таких исследованиях устанавливается тяжесть поражения чувствительности нервов конечностей. Характерный для данного заболевания электронейромиографический паттерн заключается в отсутствии или значительном снижении амплитуды потенциалов действия чувствительных нервов конечностей, при сравнительно небольшом снижении скорости проведения импульса по двигательным нервам. Для полной диагностики проводят нагрузочные тесты толерантности к глюкозе (для исключения сахарного диабета), рентгеновское исследование позвоночника. На ЭКГ - нарушение ритма, инверсия зубцаТ, изменения проводимости, при эхокарднографии особенно часто отмечаются нарушения проводимости, вплоть до полной блокады, и гипертрофия межжелудочковой перегородки. В ряде случаев клинические и электрокардиографические симптомы поражения сердца иногда на несколько лет опережают появление неврологических нарушений. Больные длительно наблюдаются у кардиолога или участкового терапевта, чаще всего с диагнозом “ревмокардит”. Для оценки митохондриальных нарушений с помощью цитохимического метода наиболее целесообразным представляется определение активности ряда ферментов-дегидрогеназ лимфоцитов: сукцинатдегидрогеназы (СДГ),
Профилактика атаксии Фридрейха - Особое значение имеет ДНК тестирование на ранней пресимптомной стадии с целью назначения превентивной терапии. Обследуются в первую очередь родственники больного.
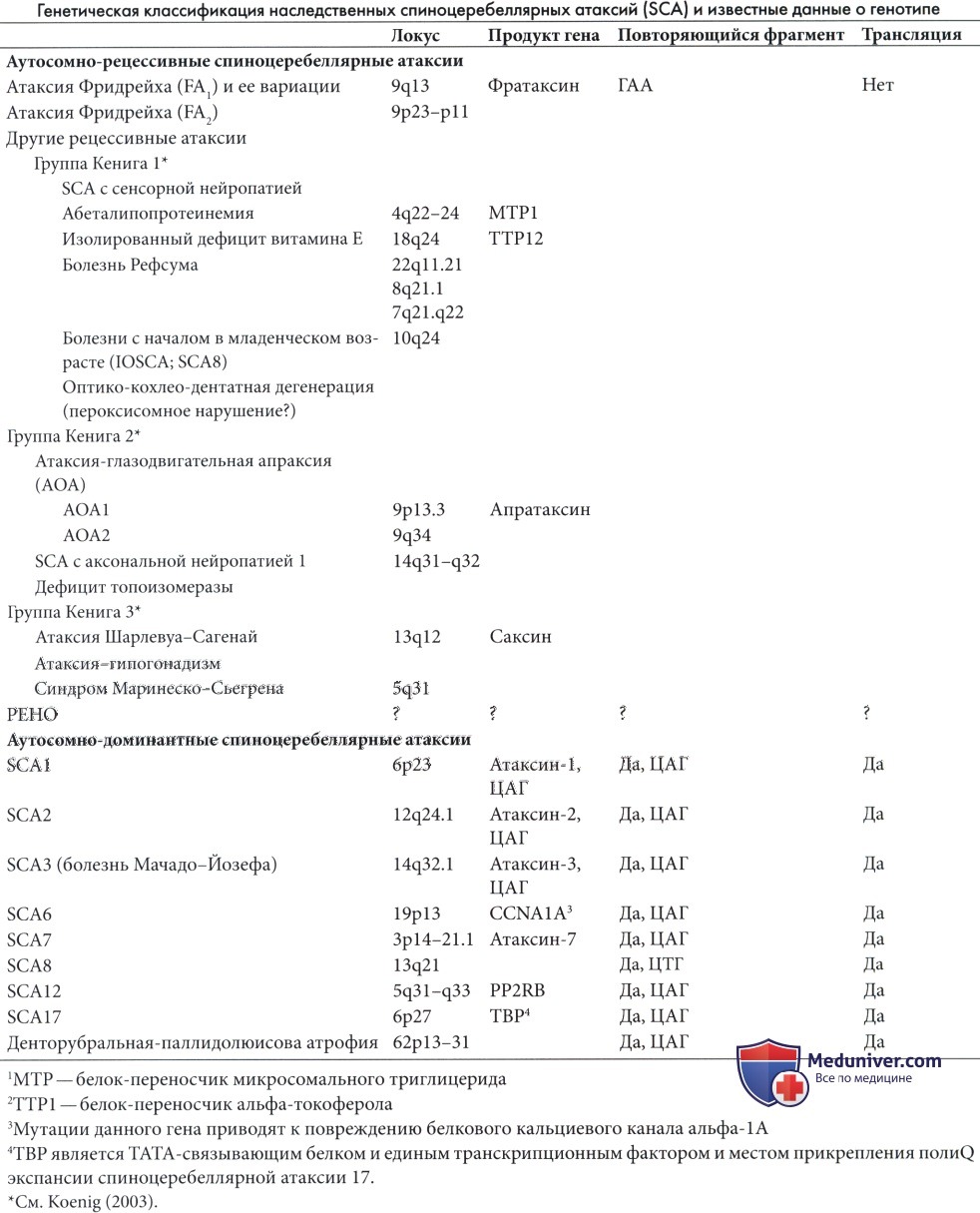
Наследуемые рецессивные спиноцеребеллярные атаксии: атаксия Фридрейха и другие
а) Атаксия Фридрейха. Атаксия Фридрейха — наиболее четко описанная и часто встречающаяся спиноцеребеллярная дегенерация. Частота встречаемости гена составляет 1:110 человек в Англии (Harding 1981a), и примерно один из 10000 человек в Швеции имеет клинические проявления.
Ген атаксии Фридрейха включает повторы ГА А последовательности в интроне 1, который распространен у пациентов (120-1700 повторов). Продуктом нормального гена является белок фратаксин, функция которого не полностью ясна. 94% пациентов с типичной атаксией Фридрейха являются гомозиготами по ГАА экспансии, тем не менее продолжительность повтора на каждой хромосоме из пары неодинакова (Durr et al., 1996a).
В редких случаях отмечается только одна мутация, но в такой ситуации выявляется точечная мутация в гомозиготном локусе (Campuzano et al., 1996). Выраженная длина повтора коррелирует с началом в раннем возрасте, более стремительным течением и наличием кардиомиопатии (Durr et al., 1996a).
Второй ген на хромосоме 9p23-p11 является причиной редких случаев (Фридрейха 2), клинически нечетко отличаемых от 1 типа (Christodoulou et al., 2001).
Сердце увеличено, и более чем в половине случаев отмечается гипертрофическая кардиомиопатия с некрозом волокон и фиброзом, преимущественно затрагивающим левый желудочек.
Критерии диагностики атаксии Фридрейха (Harding, 1981a) включают начало до 25 лет (обычно до 16 лет), аутосомно-рецессивное наследование и сочетанное поражение крупных сенсорных волокон периферических нервов, мозжечкового тракта, пирамидного тракта и задних столбов.
Тем не менее, степень фенотипической вариабельности велика, в некоторых случаях отмечается позднее начало и/или меньшая выраженность симптомов и вариабельное течение, и некоторые пациенты прикованы к инвалидной коляске в раннем подростковом возрасте, в то время как другие способны самостоятельно передвигаться почти до 40 лет (Montermini et al., 1997).
По неофициальным данным, к доминантным случаям относится большая часть наследственной моторной и сенсорной нейропатии со скелетными деформациями и утратой чувствительности, но некоторые случаи не поддаются классификации.

Клинические проявления атаксии Фридрейха были описаны у 115 пациентов из 90 семей (Harding, 1981b). Основные проявления представлены в таблице ниже.
Заболевание чаще всего начинается в возрасте 5-16 лет, в редких случаях — в возрасте 2-5 лет. Прогрессирующая атаксия нижних конечностей с нарушением походки является основным проявлением, в то время как поражение верхних конечностей, приводящее к неуклюжести, в ранние сроки отмечается только в 25% случаев.
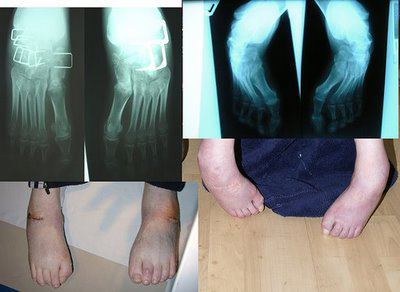
Сколиоз, тремор и изменения со стороны сердца редко являются первыми проявлениями, но формируются со временем, особенно при раннем начале заболевания. Конская стопа также является ранним симптомом. При осмотре в 70-95% случаев выявляется отсутствие глубоких рефлексов.
Дизартрия, пирамидные знаки со стороны ног и утрата глубокой и вибрационной чувствительности могут появляться позже, но относятся к постоянным симптомам. Нистагм встречается нечасто (20% случаев), медленные изломанные следящие движения глаз выявляются в 12% случаев (Harding, 1981b). Нестабильность фиксации является типичным проявлением (Alper и Narayanan, 2003).
Нередко встречается атрофия зрительного нерва, а глухота отмечается только у 10% пациентов. Дистальная атрофия отмечается практически в половине случаев. Интеллект не страдает.
Поражение сердца по результатам ЭКГ обнаруживается в трети случаев и даже чаще, если ЭКГ проводится систематически. Изменения зубца Т и аномалии сегмента ST являются ранними признаками сердечной недостаточности и единственным ее проявлением.
В конечном счете формируется прогрессирующая сердечная недостаточность или аритмия с фибрилляцией предсердий, половина пациентов умирает от сердечной недостаточности (Leone et al., 1988).
Течение заболевания медленное, но прогрессирующее. В среднем пациенты утрачивали способность ходить к 25 годам, со средней продолжительностью заболевания 15,5 лет.
Сахарный диабет является дальнейшим осложнением и развивается у 10% пациентов. Он имеет тенденцию сочетаться с атрофией зрительного нерва, в некоторых случаях диабетическая кома является причиной смерти.
Атипичные формы включают легкие случаи, которые, возможно, связаны с одним и тем же локусом 9-й хромосомы. К данной группе относятся случаи сохранения сухожильных рефлексов (Palau et al., 1995) и поздние формы с началом в раннем взрослом возрасте (De Michele et al., 1994).
Результаты одной из недавних работ, в которой использовалось возможное выявление мутантного гена, предполагают, что клиническая картина более вариабельна, чем считалось раньше (Palau et al., 1995; Pandolfo 2003). 25% пациентов в рамкам одного крупного исследования имели одно или более атипичное проявление (начало после 25 лет, сохранение или даже оживление сухожильных рефлексов или отсутствие симптома Бабинского).
Эта группа, очевидно, была гетерогенной как при раннем (
Вторая подгруппа Кенига включает атаксию со зрительной моторной апраксией (АОА), которая делится на два типа: AOA1 является одной из наиболее распространенных форм детского возраста и описана вместе с атаксией-телеангиэктазией, несмотря на то, что ее физиология кажется более сходной с спиноцеребеллярными атаксиями (SCA, см. далее). Одним из важных биологических признаков является гипоальбуминемия, которая практически постоянно обнаруживается и имеет диагностическую значимость. АОА 2 типа встречается реже и начинается позже (в позднем подростковом или раннем взрослом возрасте). Умеренно повышенный уровень альфа-фетопротеина отмечается в 75% случаев.
Клинические проявления AOA1 очень напоминают проявления атаксии-телеангиэктазии, но без признаков экстраневрологических поражений.
В отличии от атаксии-телеангиэктазии, AOA1 не связана с повышением уровня альфа-фетопротеина, хромосомными аномалиями, склонностью к раковым опухолям или повышенной радиочувствительностью культуры фибробластов (Le Ber et al., 2005). Редким, но интересным состоянием является спиноцеребеллярная атаксия с аксональной нейропатией 1 (SCA1), которая фактически является нарушением репарации ДНК, вызванной отсутствием фермента топоизомеразы-фосфодиэстеразы-1 (TDP1) (E1-Khamisy et al., 2005).
В третьей подгруппе Кенига четко описана атаксия Шарлевуа-Сагеней. Изначально синдром был описан в Квебеке, но с тех пор регистрировался и в других частях света (Gucuyener et al., 2001). Заболевание связано с мутацией гена сакцина на 13-й хромосоме (Engert et al., 2000). Фенотипические проявления включают заметную спастичность и постоянное наличие полос на глазном дне с преобладанием миелиновых волокон, радиально расходящихся от диска зрительного нерва.
Два редких аутосомно-рецессивных синдрома включают очень медленно прогрессирующую атаксию и могут рассматриваться вместе с SCA. Несмотря на то, что патология и механизмы заболеваний отличаются, они проявляются несколькими общими клиническими симптомами.
Синдром Маринеску-Шегрена включает атрофию мозжечка, преимущественно затрагивающую червь, раннее начало медленно прогрессирующей атаксии, катаракту, легкую задержку умственного развития, иногда гипогонадизм (Sewry et al., 1988) и позднее развитие специфической миопатии (Superneau et al., 1987). Заболевание развивается в результате мутации гена SLI1, кодирующего белок-шаперон, ключевой регулятор основных функций эндоплазматической сети.
Видео этиология, патогенез атаксии Фридрейха
- Вернуться в оглавление раздела "Неврология."
Редактор: Искандер Милевски. Дата обновления публикации: 12.3.2021

Доклад на СНК кафедры неврологии педиатрического факультета РГМУ 5.10.2006 года.
Болезнь Фридрейха исторически является первой нозологически самостоятельной формой наследственных атаксий, выделенной более 100 лет назад из общей группы локомоторной атаксии. Это сделал N. Friedreich . Очень компетентный врач и патолог, N.Friedreich интересовался всеми областями медицины, особенно неврологией. Friedreich обладал огромным умственным потенциалом, был одним из основоположников современной клинической неврологии.
Болезнь Фридрейха - наследственное заболевание, характеризующееся медленно прогрессирующей атаксией вследствие склеротического перерождения задних и боковых столбов спинного мозга, гипоплазией мозжечка и спинного мозга.
Болезнь Фридрейха — самая частая форма наследственных атаксий, распространенность составляет 2—7 на 100000 населения. Интересно, что данное заболевание встречается практически исключительно у лиц белой расы и не наблюдается у коренных представителей негроидной расы или азиатских народов.
Болезнь Фридрейха наследуется по аутосомно-рецессивному типу.
Ген болезни Фридрейха был картирован в центромерной области 9-й хромосомы в локусе 9ql3 — q21 (Chamberlain S. et al., 1988; Hanauer A. et al., 1990). В нашей стране первый опыт косвенной ДНК-диагностики был получен сотрудниками нейрогенетического отделения НИИ неврологии РАМН совместно с лабораторией ДНК-диагностики Медико-генетического научного центра РАМН (Пугачев В.В. и др., 1996) в 1996 г.
В 1996 г. большой международной исследовательской группой был идентифицирован новый ген FRDA (другое название Х25), мутации в котором приводят к возникновению болезни Фридрейха (Campuzano V. et al., 1996). Данный ген кодирует новый митохондриальный белок фратаксин, расположенный на внутренней поверхности мембраны митохондрий и участвующий в энергетическом метаболизме клетки (в частности, регулирующий внутриклеточный обмен железа). Сущность мутации при болезни Фридрейха заключается в экспансии тринуклеотидных повторов (т.е. патологическом увеличении числа копий).
1) участие белка в депонировании и выводе железа из митохондрий;
2) участие в синтезе Fe-S-белков;
3) защитная функция в отношении окислительного повреждения нейронов (Cavadini P. et al., 2002).
снижение уровня нормального фратаксина в тканях;
аккумуляция железа внутри митохондрий (Pandolfo M., 2001)
необратимое повреждение целостности и функции митохондрий;
нарушение процессов окислительного фосфорилирования;
Биохимические процессы в системе окислительного фосфорилирования и дыхательной цепи митохондрий являются мощным источником свободнорадикальных форм кислорода в клетке. При этом металлы переменной валентности, в том числе железо, играют важную роль в катализе реакций с образованием активных форм кислорода. Резкое усиление окислительных процессов при недостаточности или истощении системы антиоксидантной защиты приводит к развитию окислительного стресса, который в настоящее время рассматривается как один из общих механизмов повреждения тканей организма.
Таким образом, аккумуляция железа внутри митохондрий при болезни Фридрейха увеличивает продукцию свободных радикалов и митохондрии утрачивают способность эффективно осуществлять окислительное фосфорилирование. Следствием этого является снижение синтеза АТФ, а также дисфункция и необратимое повреждение клетки в условиях системного энергетического дефицита. Известно, что Fe-S-белки (в том числе аконитаза) чрезвычайно чувствительны к повреждающему действию свободных радикалов, поэтому количество их в клетке уменьшается. У больных формируется своеобразный порочный круг: окислительный стресс непосредственно вызывает недостаточность Fe-S-белков, а высвобождающееся при этом железо, в свою очередь, усиливает уже существующую перегрузку железом.
Таким образом, имеются два основных независимых друг от друга механизма клеточной токсичности при болезни Фридрейха — окислительный стресс и дефицит Fe-S-содержащих белков.
С этих позиций генетически, болезнь Фридрейха рассматривается как особая разновидность митохондриальной болезни, обусловленная повреждением ядерного гена. Патоморфологически же относится к спинальным формам наследственных атаксий.
Степень экспансии повторов в мутантном гене Болезни Фридрейха коррелирует с возрастом начала заболевания, особенностями его течения и клинической картины.
Прогрессирование патологического процесса при БФ, связано с гибелью нейронов в некоторых системах от периферии к центру, с возможным исчезновением тела клетки. Болезнь Фридрейха характеризуется дегенерацией задних и боковых столбов спинного мозга, особенно в люмбосакральных сегментах. Пучки Голля поражаются в большей степени, чем пучки Бурдаха. Происходит гибель клеток столбов Кларка и начинающихся от них дорсальных спиноцеребеллярных трактов, а также (обычно в поздней стадии болезни) дегенерацией ядер III, V, IX —X, XII пар черепных нервов, клеток Пуркинье, зубчатого ядра и верхней ножки мозжечка, изменения могут обнаруживаться и в больших полушариях мозга. Во всех этих областях выявляются аксональная дегенерация, демиелинизация и компенсаторный глиоз. Клеточную гибель и глиоз также можно заметить в вестибулярной и слуховой системах. При гистохимическом исследовании задних столбов спинного мозга определяется уменьшение гликолитических ферментов и увеличение окислительных и гидролитических. Кроме снижения уровня протеолипидов, составляющих структуру миелиновой оболочки проводящих путей, демиелинизации, патологии собственно липидов в пораженных путях не выявлено. Патология со стороны внутренних органов: кардиомегалия с гипертрофией миоцитов, а в поджелудочной железе хронический интерстициальный фиброз и воспалительная инфильтрация.
Первые симптомы заболевания возникают обычно на 1—2-м десятилетии жизни, чаще всего в препубертатном периоде, характеризуются незаметным появлением симптомов, относительно быстрым прогрессированием процесса и сочетанием типичных неврологических и экстраневральных клинических проявлений.
Типичным неврологическим проявлением болезни Фридрейха является нарушение глубокой (суставно-мышечной и вибрационной) чувствительности. Довольно рано у больных при неврологическом осмотре может быть обнаружен симптом Бабинского, мышечная гипотония. По мере прогрессирования заболевания постепенно нарастают мозжечковая и сенситивная атаксия, слабость и атрофия мышц ног. Атаксия вызвана комбинацией церебеллярной асинергии и поражением заднего столба с его чувствительными проводниками. Она обычно более выражена в ногах, чем в руках, и выявляется при исследовании походки и статики ребенка.
В поздней стадии болезни парезы, амиотрофии и расстройства глубокой чувствительности распространяются на руки. Больные перестают самостоятельно ходить и обслуживать себя из-за глубокого распада моторных функций. В ряде случаев наблюдается нистагм, снижение слуха, атрофия зрительных нервов; при длительном течении болезни отмечается нарушение функции тазовых органов. По поводу деменции мнения противоречивы. Если у взрослых она описана, то у детей умственная отсталость и деменция встречаются редко.
Патология со стороны органа зрения при атаксии Фридрейха встречается относительно редко и может включать аномалии рефракции, пигментную ретинопатию, катаракту (De Silva R. et al., 1998).
Принято считать, что экстраневральные признаки болезни Фридрейха — это проявление плейотропного действия одного мутантного гена.
- При исследовании вызванных зрительных потенциалов выявляются генерализованное снижение амплитуды потенциалов и удлинение времени их появления. Уменьшение амплитуды, вероятно, является следствием утраты функционирующих волокон в зрительном тракте. Результаты электроретинограммы обычно нормальные.
- Соматосенсорные вызванные потенциалы, регистрируемые от надключичных отведений, отличаются от нормальных уже на самых ранних стадиях болезни, но они не сопровождаются снижением проводимости по периферическому нерву.
- При молекулярно-генетическом обследовании пациентов с клинически типичными проявлениями БФ на увеличение тринуклеотида ГАА расширение аллеля обнаруживается не у всех. При этом возможна точечная мутация или делеция в гене БФ на обеих хромосомах. В этих случаях это может быть фенокопия БФ, так как увеличение триплета было описано у многих больных атипичной атаксией и у пациентов с генерализованной хореей.
- МР-томография позволяет уже в ранней стадии болезни визуализировать атрофию спинного мозга, уменьшение поперечного размера спинного мозга, особенно усиливающееся в каудальном направлении, а при более длительном течении — умеренно выраженную атрофию продолговатого мозга, моста и мозжечка.
- Компьютерная томография головного мозга имеет ограниченное значение (в связи со спинальной локализацией основных морфологических изменений). В большинстве случаев при КТ обычно обнаруживается либо слабая степень атрофии мозжечка, либо отсутствие изменений. Лишь в поздней стадии заболевания при КТ можно выявить ряд изменений: атрофию полушарий и червя мозжечка, расширение IV желудочка, стволовых цистерн, боковых желудочков и субарахноидального пространства больших полушарий.
- Электрофизиологические исследования - электронейромиографический паттерн (отсутствие или значительное снижении амплитуды потенциалов действия чувствительных нервов конечностей при сравнительно небольшом снижении скорости проведения импульса по двигательным нервам.)
- Электрокардиография и эхокардиография – выявление кардиомиопатии, (нарушение ритма, инверсия зубца Т, изменения проводимости). Особенно часто отмечаются нарушения проводимости, вплоть до полной блокады, и гипертрофия межжелудочковой перегородки. В ряде случаев клинические и электрокардиографические симптомы поражения сердца иногда на несколько лет опережают появление неврологических нарушений.
- Исследование содержания глюкозы в крови с проведением специальных нагрузочных тестов толерантности к глюкозе (для исключения сахарного диабета).
- Рентгенологическое исследование позвоночника (характеристика костных деформаций).
- Для оценки митохондриальных нарушений с помощью цитохимического метода наиболее целесообразны представляется определение активности ряда ферментов-дегидрогеназ лимфоцитов: сукцинатдегидрогеназы (СДГ),
а-глицерофосфа дегидрогеназы (ГФДГ) глутаматдегидрогеназы (ГДГ), лактатдегидрогеназы (ЛДГ), малатдегидрогеназы (МДГ) и др. Выявляется их достоверное снижение.
Основными критериями диагноза болезни Фридрейха являются:
1. аутосомно-рецессивный тип наследования;
2. дебют в подростковом, реже в юношеском возрасте;
3. атаксия, арефлексия, нарушение глубокой чувствительности, слабость и атрофии мышц ног, позднее рук;
4. экстраневральные симптомы:
б) эндокринные расстройства: сахарный диабет, гипогонадизм, инфантилизм, дисфункция яичников;
в) кардиомиопатия (гипертрофическая, реже дилатационная): изменения на ЭКГ и ЭхоКГ;
5. атрофия спинного мозга, визуализирующаяся на МР-томо-
граммах;
6. ДНК-диагностика. Диагноз подтверждается определением размера повторов ГАА.
Болезнь Фридрейха необходимо дифференцировать, в первую очередь, со второй по частоте прогрессирующей атаксией с началом в детском возрасте — атаксией-телеангиэктазией (болезнью Луи-Бар). Клинически она отличается наличием на коже телеангиэктазий (чрезмерное локальное расширение мелких сосудов, преимущественно прекапилляров и капилляров), отсутствием скелетных аномалий, частыми и тяжело протекающими инфекциями дыхательных путей, отсутствием или крайне низким уровнем IgA, высоким уровнем а-фетопротеина. На МРТ выявляется гипоплазия мозжечка, чаще его червя.
От клинически весьма сходной формы наследственной атаксии, вызываемой дефицитом витамина Е, и близким к ней синдромом Бассена—Корнцвейга. Для дифференциальной диагностики необходимо определять содержание в крови витамина Е, исследовать липидный профиль крови, мазок крови на наличие акантоцитоза.
Дифференциальный диагноз болезни Фридрейха и рассеянного склероза обычно не вызывает затруднений, поскольку для последнего нехарактерны такие симптомы, как сухожильная арефлексия, мышечная гипотония, амиотрофии, экстраневральные проявления, а также в связи с отсутствием при болезни Фридрейха ремиссий и очаговых изменений плотности вещества мозга при КТ и МР-томографии.
Дифференциальный диагноз проводят также с сифилитическим поражением ц.н.с., опухолями мозжечка, фуникулярным миелозом.
В связи с недостаточностью энергопродукции при болезни Фридрейха важное значение в лечении данного заболевания в настоящее время придается препаратам, поддерживающим функцию митохондрий и широко применяющимся в терапии других митохондриальных болезней.
Общий принцип лечения данными препаратами состоит в сочетанном назначении лекарств, синергично влияющих на разные уровни энергетического метаболизма. Рекомендуется одновременное назначение как минимум трех лекарственных средств из первых трех групп. (Препараты, повышающие активность дыхательной цепи митохондрий, кофакторы энзимных реакций энергетического обмена, антиоксиданты). Полученные предварительные результаты свидетельствуют о том, что вышеуказанный комплекс митохондриальных препаратов может способствовать определенной стабилизации энергетических процессов в клетке и тем самым в какой-то степени замедлять развитие нейродегенерации при атаксии Фридрейха.
Комплексное лечение пациентов должно включать применение довольно широкого спектра симптоматических средств. Обычно назначают препараты, улучшающие метаболизм миокарда: рибоксин, кокарбоксилазу, предуктал и др.
Дети с БФ могут оставаться активными максимально долго, занимаясь лечебной физкультурой и комплексами корректирующих упражнений, которые следует фокусировать на тренировку баланса и силы мышц. При такой программе упражнений не развивается кардиомиопатия.
Ортопедическое хирургическое лечение скелетных деформаций, особенно прогрессирующего сколиоза, который является главной причиной болей, может быть необходимым, если ношение ортопедического корсета не предотвращает развитие сколиоза. Проводится общеукрепляющее лечение (витамины), а также препараты, влияющие на тканевый обмен (пирацетам, аминалон, ацефен, церебролизин), лечение которыми следует периодически повторять.
Болезнь Фридрейха характеризуется неуклонно прогрессирующим течением, длительность болезни может варьировать в широких пределах, но чаще всего не превышает 20 лет.
Непосредственными причинами смерти могут быть сердечная и легочная недостаточность, инфекционные осложнения.
Профилактика БФ основана на медико-генетической консультации.
Болезнь Фридрейха до наших дней остается, пожалуй, наиболее четко очерченным синдромом в обширном и гетерогенном ряду атаксий; это, бесспорно, свидетельствует об исключительном таланте исследователя, именем которого названо данное заболевание.
Николаус Фридрейх умер от разорвавшейся аневризмы аорты в 1882 году в возрасте 57 лет.
Список использованной литературы
7. "Журнал неврологии и психиатрии им.С.С.Корсакова" №4 1996.
10. Журнал Science, 8.03.1996, “Friedreich’s Ataxia: Autosomal Recessive Disease Caused by an Intronic GAA Triplet Repeat Expansion.
Русская остеопатическая ассоциация - сайт доктора остеопатии Запольского Кирилла Владимировича
Атаксия Фридрейха — наследственное нейродегенеративное заболевание, для которого характерно нарушение выведения ионов железа из околомитохондриального пространства клетки.
Среди европейцев распространенность заболевания составляет 1:20 000–1:50 000, а во всем мире каждый 120-й житель имеет предрасположенность к данной патологии. Причиной развития атаксии Фридрейха является мутация в гене FXN, в частности, нестабильное увеличение триплетов GAA. Данный ген кодирует специфический белок фратаксин, который отвечает за транспорт ионов железа из околомитохондриального пространства и не допускает тем самым образования свободных радикалов, которые оказывают выраженное повреждающее действие на центральную и периферическую нервную систему, а также другие органы.
Наследование атаксии Фридрейха проходит по аутосомно-рецессивному типу. Возможно бессимптомное носительство гена.
Клинические проявления
Мутации в гене FXN не сразу приводят к выраженному клиническому проявлению атаксии Фридрейха. Заболевание может не давать о себе знать десятилетиями, но обычно первые признаки возникают в в первом десятилетии. Дебют болезни начинается с расстройств походки и координации движений. Пациент предъявляет жалобы на неуверенность, шаткость, неловкость при движениях, отмечает частые падения. Позже присоединяются расстройства движений верхних конечностей, появление тремора. Среди других проявлений атаксии Фридрейха отмечаются:
- слабость в мышцах ног;
- расстройства речи;
- снижение остроты слуха;
- исчезновение рефлексов;
- нарушение функций яичников;
- парезы и параличи;
- деменция;
- сахарный диабет;
- гипогонадизм;
- атрофия зрительного нерва.
Кроме того, заболевание сопровождается различными нарушениями работы сердца, например, аритмией, в тяжелых случаях — сердечной недостаточностью. Часто у пациентов с атаксией Фридрейха отмечаются деформации костей.
Диагностика атаксии Фридрейха
Поставить точный диагноз в некоторых случаях бывает затруднительно. Пациент может длительное время наблюдаться у невролога, кардиолога, ортопеда или других специалистов, которые не всегда могут заподозрить атаксию Фридрейха. Для того чтобы выявить характерные изменения, требуется пройти комплексное обследование, в план которого будут входить следующие методы:
- МРТ головного мозга или позвоночника;
- нейрофизиологическое тестирование;
- электронейрография;
- электромиография;
- магнитная стимуляция.
Эффективного лечения, которое смогло бы устранить причину атаксии Фридрейха, в настоящее время не разработано. Однако для улучшения качества и продолжительности жизни может применяться симптоматическая терапия, которая всегда подбирается индивидуально. Для нормализации работы митохондрий назначаются антиоксиданты, стимуляторы активности дыхательной цепи, кофакторы энзимных реакций. Деформации костей исправляются преимущественно хирургическими методами. Для коррекции эндокринных нарушений применяются гормоны.
Для того чтобы замедлить прогрессирование атаксии Фридрейха, может назначаться ЛФК, при необходимости подбираются протезы и инвалидные кресла, которые помогут пациенту сохранить активный образ жизни.
Атаксия Фридрейха является неизлечимым прогрессирующим заболеванием. Прогноз для жизни пациента во многом зависит от возраста, в котором оно развилось, и симптомов. У женщин течение более благоприятное, чем у мужчин. Наиболее опасными считаются осложнения в виде сахарного диабета, сердечной недостаточности, бронхопневмонии. При отсутствии данных нарушений пациенты могут дожить до 70 лет и более, в противном случае продолжительность жизни ограничивается 20 годами с момента начала прогрессирования болезни.
Читайте также: