
Toll like рецепторы реферат
Обновлено: 07.07.2024
Атеросклероз считается основной причиной сердечно-сосудистых заболеваний. Изучение патогенеза атеросклероза является важнейшим научно-исследовательским направлением. В патогенезе атеросклероза принимают участие множество механизмов, среди которых наиболее значимыми являются нарушения обмена липидов и иммуновоспалительные процессы. В последнее время активно изучается роль механизмов врожденной иммунной системы в патогенезе атеросклероза. Одними из ключевых компонентов врожденной иммунной системы являются Толл-подобные рецепторы (TLR). К настоящему времени было идентифицировано более 10 разновидностей TLR. TLR экспрессируются во многих клетках, включая иммунные (макрофаги, дендритные клетки) и неиммунные клетки (эпителиальные). Основная функция TLR заключается в распознавании патоген-ассоциированных молекулярных паттернов. Многочисленные данные свидетельствуют о том, что TLR являются одними из ключевых организаторов иммуновоспалительного процесса при атеросклерозе. Среди TLR наиболее изученными участниками патогенеза атеросклероза являются TLR2 и TLR4. Во многих исследованиях показано, что повышение экспрессии TLR2 и TLR4 усиливает атеросклероз, а ингибирование, напротив, ослабляет. В настоящей статье суммированы данные о роли TLR в патогенезе атеросклероза, обсуждаются возможности использования TLR в качестве мишеней для терапевтического воздействия при атеросклерозе.

1. Чаулин А.М., Карслян Л.С., Григорьева Е.В., Нурбалтаева Д.А., Дупляков Д.В. Клинико-диагностическая ценность кардиомаркеров в биологических жидкостях человека // Кардиология. 2019. Т. 59. № 11. С. 66–75.
2. Куранов А.А., Балеев М.С., Митрофанова Н.Н., Мельников В.Л. Некоторые аспекты патогенеза атеросклероза и факторы риска развития сердечно-сосудистых заболеваний // Фундаментальные исследования. 2014. № 10–6. С. 1234–1238.
5. Libby P., Okamoto Y., Rocha V.Z., Folco E. Inflammation in atherosclerosis: transition from theory to practice. Circ. J. 2010. Vol. 4. № 2. P. 213–220.
6. Чаулин А.М. Участие пропротеинконвертазы субтилизин кексин типа 9 в патогенезе атеросклероза // Известия высших учебных заведений. Поволжский регион. Медицинские науки. 2020. Vol. 1. № 53. P. 111–128.
7. Чаулин А.М., Григорьева Ю.В., Дупляков Д.В. Современные представления о патофизиологии атеросклероза. Ч. 1. Роль нарушения обмена липидов и эндотелиальной дисфункции (обзор литературы) // Медицина в Кузбассе. 2020. № 2. С. 34–41.
8. Newton K., Dixit V.M. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol. 2012. Vol. 4. № 3. P. a006049.
9. Andersson U., Wang H., Palmblad K., Aveberger A.C., Bloom O., Erlandsson-Harris H., Janson A., Kokkola R., Zhang M., Yang H., Tracey K.J. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J. Exp. Med. 2000. Vol. 192. № 4. P. 565–570.
10. Seneviratne A.N., Sivagurunathan B., Monaco C. Toll-like receptors and macrophage activation in atherosclerosis. Clin Chim Acta. 2012. Vol. 413. № 1–2. P. 3–14.
11. Akira S., Uematsu S., Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006. Vol. 124. № 4. P. 783–801.
12. Cole J.E., Mitra A.T., Monaco C. Treating atherosclerosis: the potential of Toll-like receptors as therapeutic targets. Expert Rev Cardiovasc Ther. 2010. Vol. 8. № 11. P. 1619–1635.
13. Stewart C.R., Stuart L.M., Wilkinson K., van Gils J.M., Deng J., Halle A., Rayner K.J., Boyer L., Zhong R., Frazier W.A., Lacy-Hulbert A., El Khoury J., Golenbock D.T., Moore K.J. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010. Vol. 11. № 2. P. 155–161.
14. Takeda K., Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005. Vol. 17. № 1. P. 1–14.
16. Коровкина Е.С., Кажарова С.В. Роль Toll-подобных рецепторов в патогенезе воспалительных заболеваний бронхолегочной системы // Инфекция и иммунитет. 2016. Т. 6. № 2. С. 109–116.
17. Щебляков Д.В., Логунов Д.Ю., Тухватулин А.И., Шмаров М.М., Народицкий Б.С., Гинцбург А.Л. Тoлл-подобные рецепторы (TLR) и их значение в опухолевой прогрессии // Acta Naturae (русскоязычная версия). 2010. Т. 2. № 3 (6). С. 28–37.
18. Dunzendorfer S., Lee H.K., Tobias P.S. Flow-dependent regulation of endothelial Toll-like receptor 2 expression through inhibition of SP1 activity. Circ Res. 2004. Vol. 95. № 7. P. 684–691.
19. Mullick A.E., Tobias P.S., Curtiss L.K. Modulation of atherosclerosis in mice by Toll-like receptor 2. J. Clin. Invest. 2005. Vol. 115. № 11. P. 3149–3156.
20. Mullick A.E., Soldau K., Kiosses W.B., Bell T.A. 3rd., Tobias P.S., Curtiss L.K. Increased endothelial expression of Toll-like receptor 2 at sites of disturbed blood flow exacerbates early atherogenic events. J. Exp Med. 2008. Vol. 205. № 2. P. 373–383.
21. Madan M., Amar S. Toll-like receptor-2 mediates diet and/or pathogen associated atherosclerosis: proteomic findings. PLoS One. 2008. Vol. 3. № 9. P. e3204.
22. Monaco C., Gregan S.M., Navin T.J., Foxwell B.M., Davies A.H., Feldmann M. Toll-like receptor-2 mediates inflammation and matrix degradation in human atherosclerosis. Circulation. 2009. Vol. 120. № 24. P. 2462–2469.
23. Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol. 2005. Vol. 25. № 11. P. 2255–2264.
24. Seimon T.A., Nadolski M.J., Liao X., Magallon J., Nguyen M., Feric N.T., Koschinsky M.L., Harkewicz R., Witztum J.L., Tsimikas S., Golenbock D., Moore K.J., Tabas I. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. 2010. Vol. 12. № 5. P. 467–482.
25. Higashimori M., Tatro J.B., Moore K.J., Mendelsohn M.E., Galper J.B., Beasley D. Role of toll-like receptor 4 in intimal foam cell accumulation in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2011. Vol. 31. № 1. P. 50–57.
26. Curtiss L.K., Black A.S., Bonnet D.J., Tobias P.S. Atherosclerosis induced by endogenous and exogenous toll-like receptor (TLR)1 or TLR6 agonists. J. Lipid Res. 2012. Vol. 53. № 10. P. 2126–2132.
27. Michelsen K.S., Wong M.H., Shah P.K., Zhang W., Yano J., Doherty T.M., Akira S., Rajavashisth T.B., Arditi M. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci U S A. 2004. Vol. 101. № 29. P. 10679–10684.
28. Xu X.H., Shah P.K., Faure E., Equils O., Thomas L., Fishbein M.C., Luthringer D., Xu X.P., Rajavashisth T.B., Yano J., Kaul S., Arditi M. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation. 2001. Vol. 104. № 25. P. 3103–3108.
29. Dasu M.R., Devaraj S., Park S., Jialal I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care. 2010. Vol. 33. № 4. P. 861–868.
30. Bomfim G.F., Dos Santos R.A., Oliveira M.A., Giachini F.R., Akamine E.H., Tostes R.C., Fortes Z.B., Webb R.C., Carvalho M.H. Toll-like receptor 4 contributes to blood pressure regulation and vascular contraction in spontaneously hypertensive rats. Clin Sci (Lond). 2012. Vol. 122. № 11. P. 535–543.
31. Macritchie N., Grassia G., Sabir S.R., Maddaluno M., Welsh P., Sattar N., Ialenti A., Kurowska-Stolarska M., McInnes I.B., Brewer J.M., Garside P., Maffia P. Plasmacytoid dendritic cells play a key role in promoting atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2012. Vol. 32. № 11. P. 2569–2579.
32. Subramanian M., Thorp E., Hansson G.K., Tabas I. Treg-mediated suppression of atherosclerosis requires MYD88 signaling in DCs. J. Clin. Invest. 2013. Vol. 123. № 1. P. 179–188.
33. Bertocchi C., Traunwieser M., Dörler J., Hasslacher J., Joannidis M., Dunzendorfer S. Atorvastatin inhibits functional expression of proatherogenic TLR2 in arterial endothelial cells. Cell Physiol Biochem. 2011. Vol. 28. № 4. P. 625-630.
34. Arslan F., Smeets M.B., O’Neill L.A., Keogh B., McGuirk P., Timmers L., Tersteeg C., Hoefer I.E., Doevendans P.A., Pasterkamp G., de Kleijn D.P. Myocardial ischemia/reperfusion injury is mediated by leukocytic toll-like receptor-2 and reduced by systemic administration of a novel anti-toll-like receptor-2 antibody. Circulation. 2010. Vol. 121. № 1. P. 80–90.
35. Arslan F., Keogh B., McGuirk P., Parker A.E. TLR2 and TLR4 in ischemia reperfusion injury. Mediators Inflamm. 2010. Vol. 2010. P. 704202.
36. Arslan F., Houtgraaf J.H., Keogh B., Kazemi K., de Jong R., McCormack W.J., O’Neill L.A., McGuirk P., Timmers L., Smeets M.B., Akeroyd L., Reilly M., Pasterkamp G., de Kleijn D.P. Treatment with OPN-305, a humanized anti-Toll-Like receptor-2 antibody, reduces myocardial ischemia/reperfusion injury in pigs. Circ Cardiovasc Interv. 2012. Vol. 5. № 2. P. 279–287.
37. Ta N.N., Schuyler C.A., Li Y., Lopes-Virella M.F., Huang Y. DPP-4 (CD26) inhibitor alogliptin inhibits atherosclerosis in diabetic apolipoprotein E-deficient mice. J. Cardiovasc Pharmacol. 2011. Vol. 58. № 2. P. 157–166.
38. Lu Z., Zhang X., Li Y., Jin J., Huang Y. TLR4 antagonist reduces early-stage atherosclerosis in diabetic apolipoprotein E-deficient mice. J. Endocrinol. 2013. Vol. 216. № 1. P. 61–71.
Атеросклероз вызывает сердечно-сосудистые заболевания (ССЗ), которые являются ведущей причиной смертности и инвалидизации населения во всем мире. В связи с этим изучение патофизиологии, поиск новых биомаркеров для ранней диагностики атеросклероза и ССЗ, а также новых мишеней для терапевтического воздействия является приоритетной задачей современного здравоохранения [1–4].
На данный момент известно, что в патогенезе атеросклероза принимают участие многочисленные механизмы, в числе которых особенно стоит отметить нарушение обмена липидов и иммуновоспалительные процессы [4–6]. Иммуновоспалительные процессы тесно связаны с нарушением обмена липидов [7]. В последнее время появляется все больше доказательств, что механизмы врожденного иммунитета могут инициировать и ускорять атеросклероз. Так, некоторые исследования связывают патогенез атеросклероза с активацией сигнальных путей врожденного иммунитета [5]. Одними из основных компонентов системы врожденного иммунитета являются Толл-подобные рецепторы (TLR).
Цель нашего обзора заключается в описании роли TLR в патогенезе атеросклероза. По ходу статьи мы последовательно рассмотрим структуру, функционирование и основные разновидности TLR, роль TLR в развитии атеросклероза, а также обсудим возможности использования TLR в качестве мишеней для воздействия терапевтических средств с целью лечения атеросклероза.
TLR: ключевые паттерн-распознающие рецепторы (PRR)
Врожденная иммунная система поддерживает наблюдение за экзогенными патогенами или повреждением клеток при помощи поверхностно-экспрессированных паттерн-распознающих рецепторов или образ-распознающих рецепторов (англ. pattern recognition receptors, PRR) [8]. PRR расположены на поверхности клетки и в цитоплазме, где они обнаруживают патоген-ассоциированные молекулярные паттерны (англ. pathogen-associated molecular pattern PAMP), такие как липополисахарид, высвобождаемые грамотрицательными бактериями, или вирусные РНК [8]. Считается также, что эти рецепторы также распознают молекулярные фрагменты, ассоциированные с повреждением (англ. damage-associated molecular pattern, DAMP), такие как высокомобильный белок box 1 (HMGB1) или амфотерин, секретируемый иммунными клетками при воздействии провоспалительных сигналов [9]. Семейство PRR состоит из TLR, рецепторов, подобных индуцируемой ретиновой кислотой генам I (RIG-I / RLR), рецепторов, подобных домену нуклеотид-связывающей олигомеризации (NOD-like receptors, NLR), и рецепторов лектинов C-типа (CLR) [10].
TLR являются наиболее хорошо охарактеризованными PRR, из которых 11 были идентифицированы у людей и 13 у мышей [11]. TLR экспрессируются рядом иммунных клеток, такими как макрофаги, дендритные клетки и неиммунными клетками, такими как эпителиальные клетки [11]. Такие разновидности TLR, как TLR1, TLR2, TLR4, TLR5, TLR6 и TLR11, экспрессируются во внеклеточном пространстве и распознают липопептиды [12]. Бактериальные и вирусные нуклеиновые кислоты распознаются TLR3, TLR7, TLR8, TLR9 и мышиным TLR13. Все они находятся в эндоплазматическом ретикулуме, эндосомах и лизосомах.
Клиническое значение TLR и его сигнальных путей огромное. Показано, что TLR принимают участие в патогенезе многих заболеваний, в частности инфекционных [15], воспалительных [16], онкологических [17], атеросклероза и ССЗ и других. Ниже мы более подробно рассмотрим роль TLR в патогенезе атеросклероза и ССЗ. Что касается патогенеза атеросклероза, то на данный момент наиболее изучен вклад TLR2 и TLR4.
TLR при атеросклерозе
Некроз бляшек, как известно, возникает в результате апоптоза макрофагов при запущенных (осложненных) атеросклеротических поражениях [23]. Исследователь T. Seimon с соавт. показали, что окисленные фосфолипиды, окисленные липопротеины низкой плотности, насыщенные жирные кислоты и липопротеин (а) запускают апоптоз в макрофагах посредством механизмов, требующих участия TLR2 и CD36. Гибель макрофагов приводит к росту некротического ядра бляшки [24]. В исследовании M. Higashimori с коллегами обнаружено, что дефицит TLR2 снижает накопление пенистых клеток в подверженных поражению участках аорты у мышей ApoE-/- [25], что дополнительно подтверждает выводы, полученные в исследовании A. Mullick et al. [20]. В другом исследовании дефицит TLR6 или TLR1 не уменьшал атеросклероз, вызванный диетой с высоким содержанием жиров [26], а это свидетельствует о том, что TLR6 и TLR1 по отдельности не могут значимым образом влиять на атерогенез, но, очевидно, будут действовать вместе с TLR2 как гетеродимеры [26].
У ApoE-/- мышей, нокаутированных по TLR4, отмечалось снижение развития атеросклеротических поражений; кроме того, моноциты инфильтрировали очаг атеросклеротического поражения в меньшей степени у Apo -/- TLR4-/- по сравнению с ApoE-/- мышами [19, 27]. Увеличение экспрессии TLR4 наблюдалось при атеросклерозе после стимуляции окисленными липопротеинами низкой плотности [28]. Генетическая делеция TLR4 уменьшает атерогенез и инфильтрацию моноцитов/макрофагов, что также сопровождается снижением уровней ИЛ-12 и моноцитарного хемоаттрактантного протеина-1 [27]. О важной роли TLR2 и TLR4 в регуляции апоптоза макрофагов и площади некротического ядра атеросклеротической бляшки сообщается также в исследованиях T. Seimon et al. [24] и L. Curtiss et al. [26].
TLR также тесно связаны с некоторыми факторами риска развития атеросклероза. Одними из наиболее важных факторов риска являются метаболические заболевания, включая сахарный диабет 2-го типа и артериальную гипертензию. Люди с диабетом 2-го типа имеют множество метаболических отклонений, и воспалительные процессы у них протекают активнее, что увеличивает риск развития атеросклероза и ССЗ [29]. В исследовании M. Dasu с соавт. изучалась экспрессия TLR2 и TLR4 в моноцитах пациентов с сахарным диабетом 2-го типа. Показано, что у субъектов с диабетом 2-го типа экспрессия TLR2 и TLR4 в моноцитах была достоверно выше, чем у контрольных пациентов (p κ B и ряда провоспалительных цитокинов, включая ИЛ-1 и ИЛ-6 [29]. На основании полученных результатов исследователи пришли к выводу, что повышенная экспрессия TLR2 и TLR4 может вносить важный вклад в провоспалительное состояние и повышение риска развития атеросклероза и ССЗ. В другом исследовании изучалась связь TLR с артериальной гипертензией. Показано, что TLR4 участвует в регуляции артериального давления и сократимости сосудов. При введении ингибиторов TLR4 у крыс снижалось артериальное давление и сократимость сосудов, а также происходило снижение экспрессии фермента циклооксигеназы-2 (ЦОГ-2) и провоспалительного цитокина ИЛ-6 [30].
TLR как перспективные мишени для терапевтического воздействия при атеросклерозе
Функциональный анализ эндартерэктомий сонных артерий человека показал, что блокада TLR2 может оказывать благоприятное воздействие на течение атеросклероза, опосредованное ингибированием продукции провоспалительных цитокинов, хемокинов и ММП, а также ослаблением активности ядерного NF- κ B [22]. Показано, что снижение экспрессии TLR2 при лечении аторвастатином вызывает антиатеросклеротический эффект в человеческих эндотелиальных клетках артерий [32]. Исследователи F. Arslan с соавт. продемонстрировали, что моноклональные антитела против TLR2 (препарат OPN-301) приводят к уменьшению инфильтрации нейтрофилов, макрофагов и Т-лимфоцитов, а также к снижению продукции провоспалительных ФНО-α, ИЛ-1α и ГМ-КСФ на мышиной модели [33]. Несколько позже F. Arslan et al. cообщили о создании первого гуманизированного антитела против TLR2 (препарат OPN-305), которое уменьшало размер инфаркта, сохраняло систолическую функцию и в конечном итоге предотвращало повреждение миокарда на экспериментальной модели ишемии/реперфузии у свиней [34, 35]. Другими исследователями в эксперименте на мышах показано, что ингибирование TLR4-опосредованной экспрессии провоспалительных цитокинов уменьшает размер атеросклеротических поражений у мышей с сахарным диабетом [36]. Показано, что антагонист TLR4 предотвращает экспрессию проатерогенных/провоспалительных факторов ИЛ-6 и ММП-9, накопление макрофагов в атеросклеротических бляшках и активность NF- κ B у ApoE -/- мышей [37]. Существующие до сих пор терапевтические возможности для нацеливания на TLR довольно ограничены, и необходимы дальнейшие доклинические разработки. Показано, что блокировка TLR2 и TLR4 является весьма перспективной в плане разработки препаратов для клинических испытаний. По мере разработки новых инструментов для блокирования различных TLR будет получено больше доказательств возможности и эффективности их блокады при атеросклерозе и ССЗ.
Таким образом, согласно представленным выше сведениям, TLR играют важную роль в патогенезе атеросклероза, связывая механизмы врожденной иммунной защиты с воспалительными процессами при атеросклерозе. На данный момент хорошо изучена роль TLR2 и TLR4 в патогенезе атеросклероза. TLR2 и TLR4 представляет собой многообещающую мишень для воздействия терапевтических агентов при атеросклерозе. Дальнейшие исследования, способствующие пониманию патофизиологии атеросклероза, позволят разработать новые стратегии для борьбы с этим заболеванием в ближайшие годы.

Обзор
Чарльз Джейнуэй-младший (1943–2003) (слева) и Руслан Меджитов, профессор иммунологии в Йельском университете, исследователь Медицинского института Говарда Хьюза (справа)
Автор
Редакторы
Осенью прошлого года я была ошеломлена, когда не увидела имени Руслана Меджитова среди лауреатов Нобелевской премии по физиологии и медицине 2011 года. Его вклад в открытие толл-подобных рецепторов широко признан: в 2010 году он разделил с Жюлем Хоффманом Премию Розенстила за разъяснение механизмов врождённого иммунитета и был среди лауреатов премии Шао в 2011-м. Чарльз Джейнуэй предложил революционную идею: толл-подобные рецепторы врождённого иммунитета активируют адаптивный иммунный ответ. Руслан Меджитов первым открыл толл-подобный рецептор позвоночных и подтвердил идею Джейнуэя экспериментально. Протест против решения Нобелевского комитета, который не упоминает о вкладе Чарльза Джейнуэя и Руслана Меджитова, уже выразили и известные иммунологи, и студенты.
В 80-х годах прошлого столетия в центре внимания иммунологов была адаптивная (или специфическая) иммунная система. Специфический иммунный ответ (например, продукция антител против конкретного вируса) адаптируется к разнообразию патогенов, с которыми организм сталкивается в течение жизни, и формирует иммунологическую память. Большинство иммунологов изучало молекулярные детали рецепторов Т- и В-лимфоцитов и эффекторных механизмов адаптивного иммунитета (таких как антитела и лимфоциты-киллеры). Было известно, что репертуар рецепторов на поверхности лимфоцитов до встречи с антигеном не лимитирован и способен узнать практически любую молекулу [1].
Механизмы врождённого иммунитета (например, функции макрофагов и нейтрофилов) также изучались, но их рассматривали отдельно от адаптивного иммунитета, не предполагая существования связи между ними. Рецепторы клеток врождённого иммунитета генетически закодированы и неизменны в течение жизни.
Революционная идея Чарльза Джейнуэя
Некоторые экспериментальные факты, известные к тому времени, нельзя было объяснить, если рассматривать адаптивный иммунитет изолированно.
Паттерн-распознающие рецепторы передают сигнал о присутствии патогенов в организме. Джейнуэй предположил, что эти рецепторы генетически закодированы и должны узнавать жизненно важные для микробов молекулы, которые не могут быть изменены в результате одной мутации. Возможные кандидаты — сложные углеводы клеточной стенки или липопротеины. Такие молекулы входят в состав адъювантов, механизм действия которых до этого был совершенно непонятен. Теория Джейнуэя объясняла, что молекулы микробов, входящие в состав адъювантов, взаимодействуют с паттерн-распознающими рецепторами, имитируя таким образом инфекцию и активируя антиген-презентирующие клетки.

Рисунок 1. Адаптивный иммунный ответ не индуцируется, когда нет костимулирующего сигнала (слева). Липополисахарид микробов (ЛПС) активирует толл-подобные рецепторы (ТПБ-4) и индуцирует сигнал 2 (справа), вызывая размножение лимфоцитов. В этом случае белки главного комплекса гистосовместимости (ГКГ) презентируют пептиды рецепторам Т- и B-лимфоцитов (сигнал 1) при участии костимулирующей молекулы B7.
Джейнуэй провёл параллели между эффекторными механизмами врождённой и адаптивной иммунных систем. Он предположил, что паттерн-распознающие рецепторы существовали задолго до возникновения адаптивного иммунитета и активировали неспецифический иммунный ответ у беспозвоночных, у которых адаптивный иммунитет отсутствует [3]. В рамках теории Джейнуэя адаптивная иммунная система стала рассматриваться как дополнение, обеспечивающее специфичность распознавания, к эволюционно более древней врождённой иммунной системе.
Поиск паттерн-распознающих рецепторов
Руслан Меджитов в лаборатории Чарльза Джейнуэя сконцентрировался на поиске рецепторов на поверхности антиген-презентирующих клеток, которые способны распознавать микробные молекулярные паттерны (чаще всего это углеводы) и активировать костимуляторные молекулы [6]. Внеклеточный домен таких рецепторов должен узнавать углеводы. Белки, высокоспецифично связывающие углеводные остатки, называются лектинами; наиболее изученный иммунологами представитель этого семейства — лектин, связывающий маннозу. Его лектиновый домен типа C (кальций (Cа)-зависимый) был наиболее вероятным внеклеточным доменом нового рецептора [6]. Внутриклеточный домен нового рецептора должен передавать сигнал для активации костимулирующих молекул и воспалительных цитокинов. Рецептор, узнающий липополисахарид, был неизвестен, но было ясно, что он активирует сигнальный путь, ведущий к транскрипционному фактору NF-κB. Этот транскрипционный фактор является ключевым сигнальным компонентом врождённого иммунитета, вероятно также необходимым для активации костимулирующих молекул. Один из хорошо изученных рецепторов, активирующих NF-κB (рецептор интерлейкина 1), содержит внутриклеточный TIR домен, гомологичный такому же домену толл-рецептора дрозофилы. Предположение Руслана Меджитова, что новый рецептор также может содержать внутриклеточный TIR домен [6], впоследствии окажется верным. Итак, Р. Меджитов искал новый трансмембранный белок, содержащий внеклеточный лектиновый домен и внутриклеточный TIR домен [6].
В начале 1996 года он нашёл в базе данных экспрессируемых последовательностей ДНК последовательность, кодирующую белок, который имел сходство с TIR доменом; внеклеточный домен этого белка также имел сходство с толл-рецептором дрозофилы и не содержал лектинового домена. Теперь этот белок известен как толл-подобный рецептор 4.
Толл-рецептор дрозофилы

Рисунок 2. Жюль Хоффманн, руководитель научно-исследовательских работ французского Национального центра научных исследований в Страсбурге, президент Французской Академии наук
Отсутствие лектинового внеклеточного домена у толл-подобного рецептора 4 и его гомология с внеклеточным доменом толл-рецептора дрозофилы сначала вызвали разочарование. Дело в том, что известный лиганд толл-рецептора был эндогенным и никак не был связан с распознаванием микробов. Толл-рецептор, открытый в 1988 году [7], активируется во время эмбриогенеза, и его роль в иммунной системе была неизвестна.
В том же 1996 году Жюль Хоффманн (J. Hoffmann) опубликовал своё открытие: сигнальный путь толл-рецептора необходим для защиты дрозофилы от грибковых инфекций [8].
Профессора Хоффманна интересовало, как активируется экспрессия антимикробных пептидов [20] (одного из эффекторных механизмов врождённого иммунитета) у дрозофилы. Он заметил поразительное сходство между транскрипционным фактором NF-κB позвоночных и морфогенным транскрипционным фактором Dorsal у дрозофилы [8]. Dorsal активен во время эмбрионального развития, но сигнальный путь, ведущий к его активации, очень похож на сигнальный каскад, активный во время воспалительного ответа у позвоночных.
Жюль Хоффманн и его коллеги показали, что дрозофилы с мутацией в сигнальном пути толл-рецептора подвержены грибковым инфекциям, и что этот рецептор необходим для индукции противогрибкового пептида [8].
Это известие укрепило предположение Руслана Меджитова, что открытый им человеческий толл-подобный рецептор 4 и есть новый паттерн-распознающий рецептор, который активирует адаптивный иммунный ответ.
Экспериментальное подтверждение теории Джейнуэя
Чтобы подтвердить своё предположение экспериментально, Руслан Меджитов создал мутантный толл-подобный рецептор 4, находящийся в активированном состоянии постоянно (поскольку его природный лиганд был неизвестен). Клонирование человеческого толл-рецептора было описано в блестящей статье в Nature в 1997 году [9]. Этот рецептор индуцировал экспрессию костимулирующей молекулы В7 и воспалительных цитокинов, которые необходимы для активации наивных Т-лимфоцитов [9].
Эксперименты, описанные в этой статье, доказывали существование паттерн-распознающих рецепторов у позвоночных, активация которых приводит к инициации адаптивного иммунного ответа. Основное положение теории Джейнуэя было подтверждено экспериментально [10]. Это открытие привело к взрыву интереса иммунологов к врождённому иммунитету и заложило основу для интерпретации последующих экспериментов.
Дальнейшее развитие: лиганды для толл-подобных рецепторов
В течение последующих нескольких лет стало ясно, что толл-подобные рецепторы позвоночных узнают микробные компоненты непосредственно (в отличие от толл рецептора дрозофилы). Годовский (P.J. Godowsky) и коллеги первыми показали, что экстракт ЛПС, содержащий бактериальные липопротеины, активирует толл-подобный рецептор 2 [11]. Сейчас известно, что именно липопротеины являются лигандами этого рецептора.
В распознавании ЛПС участвует не один, а несколько белков, и поэтому выяснение этого механизма было нелёгким. Ещё с 1960-х годов известны линии мышей, нечувствительных к ЛПС. В 1998 году Джерард (C. Gerard) обратил внимание, что ген, кодирующий толл-подобный рецептор 4, находится в локусе, мутированном у мышей, нечувствительных к ЛПС [12]. Бётлер (B. Beutler) в 1998 году [13] и Мало (D. Malo) в 1999 году [14] методом позиционного клонирования подтвердили, что мутации именно в открытом Русланом Меджитовым гене, кодирующем толл-подобный рецептор 4, вызывают у этих мышей нечувствительность к ЛПС. Мияке (K. Miyake) и коллеги открыли недостающий компонент рецептора к ЛПС — МД-2 в 1999 году [15].
В настоящее время известно несколько классов паттерн-распознающих рецепторов, как мембранных, так и цитоплазматических [16].
Нобелевская премия по физиологии и медицине 2011 года

Полторак А. Н. Toll-подобные рецепторы как парадигма клетки // Journal of Biomedical Technologies. 2014. № 1. С. 52–57.

Распознавание патогенов микробного происхождения является основополагающим компонентом иммунного ответа, включающим воспаление (Janeway, 1998). Этот ответ опосредуется рецепторами особого семейства, узнающими наиболее общие молекулярные компоненты (паттерны, PAMP – Pathogen Associated Molecular Patterns) микробов (вирусов, бактерий, паразитов и т.д.) и получившими название PRR (Pattern Recognition Receptors) (Medzhitov, 2000). После узнавания соответствующего специфического паттерна PRR запускают серию сигнальных каскадов, которые представляют собой первую линию защиты от микробов. Кроме того, инициируемый PRR сигнал запускает созревание дендритных клеток, которые подготавливают вторую линию иммунного ответа на инфекцию, известную как приобретенный иммунитет.
Toll-подобные рецепторы (TLR) были первыми идентифицированными PRRs (Рисунок) (Lemaitre, 1996; Medzhitov, 1997).
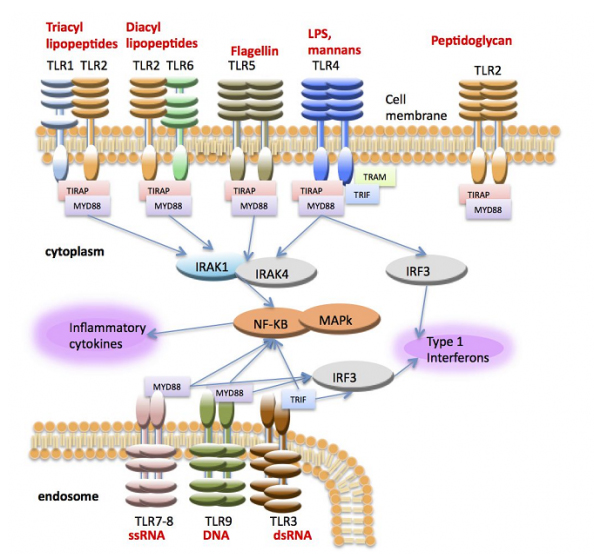
Рисунок. Активация воспалительных цитокинов и интерферона через TLRs начинается с распознавания TLR соответствующих агонистов (красный цвет). Взаимодействие адаптеров ТLRs и киназ (IRAK 1-4) приводит к активации NF-kB и MAP-киназ, основных регуляторов транскрипции цитокинов и интерферона (Lemaitre, 1996; Medzhitov, 1997).
Figure. Activation of production of inflammatory cytokines and interferon through TLRs is triggered by recognition of agonists by TLR (red). Interaction of TLRs adaptors with kinases (IRAK 1-4) leads to activation NF-kB and MAP-kinases, which are the main regulators of transcription of cytokines and interferon (Lemaitre, 1996; Medzhitov, 1997).
Для TLRs также наиболее подробно изучен и идентифицирован спектр PAMPs. TLRs – это трансмембранные белки первого типа, содержащие лейцин-богатые повторы в эктодомене, трансмембранный и цитозольные домены. Эктодомен узнает микробные паттерны, в то время как цитозольный домен активирует сигнальные пути в цитоплазме клетки. В зависимости от локализации TLRs в клетке выделяют рецепторы, расположенные в цитоплазматической мембране (TLR1, TLR2, TLR4, TLR5, TLR6, TLR10 и TLR11) или в мембранах внутриклеточных органелл (TLR3, TLR7, TLR8 и TLR9) — лизосом, эндосом, аппарата Гольджи. Лигандами рецепторов, локализованных на цитоплазматической мембране, являются поверхностные структуры микроорганизмов — липопротеин, липополисахариды, флагеллин, зимозан. Рецепторы, локализованные в мембранах внутриклеточных органелл, распознают молекулы ядерных структур микроорганизмов, но могут быть активированы и поврежденными молекулярными структурами собственного организма. Каждый TLR обнаруживает различные PAMPs вирусного, бактериального и другого происхождения. Так, например, липопротеины узнаются различными гетеродимерами из TLR1, TLR2 и TLR6 (Takeuchi, 1999), двуцепочечная РНК (DS) - TLR3 (Alexopoulou, 2001), одноцепочечная РНК (SS) - TLR7 и TLR8 (Hemmi, 2002; Heil, 2003), флагеллин - TLR5 (Smith, 2003), ДНК - TLR9 (Kumagai, 2008) (Таблица). TLR4 был первым рецептором с идентифицированным лигандом - бактериальным липополисахаридом (ЛПС) (Poltorak, 1998). В настоящее время у человека и мыши идентифицированы 10 и 12 функциональных TLR, соответственно.
Таблица. Toll-подобные рецепторы и их лиганды
В состоянии покоя неактивированные TLRs находятся на мембране клеток в мономерной форме. После активации Toll-подобных рецепторов происходит их олигомеризация. Олигомерный рецептор способен связывать несколько внутриклеточных адаптерных белков, которые обеспечивают последующую передачу сигнала. Эти белки имеют участок специфического связывания с активированными Toll-подобными рецепторами, TIR (от англ. Toll-interleukin-1 receptor) домен, который состоит из 3 консервативных участков, участвующих в белок-белковом взаимодействии. Всего существует 5 адаптерных белков с TIR-доменом: MyD88, TIRAP, TRIF, TRAM и SARM. Различные рецепторы имеют свой набор этих адаптерных белков, необходимых для передачи сигнала. Только рецептор TLR4 способен связывать все 5 белков.
Инициированная TLRs схема передачи сигнала внутрь клетки оставляет открытыми много вопросов. В частности, каким образом активация одного и того же Тoll-рецептора в разных клетках приводит к продукции совершенно разного спектра цитокинов и других воспалительных белков. Например, известно, что TLR9 в макрофагах приводит к продукции классических цитокинов, таких как TNF и IL6, в то время как тот же TLR9 в плазмацитоидных дендритных клетках приводит к продукции IFN-I (Uematsu, 2005). Аналогично, будучи локализованным на клеточной мембране, TLR4 активирует классические цитокины, а транслокация в эндосому переключает TLR4 на продукцию типа 1 интерферона (Kagan, 2008). Только одним клеточным окружением трудно объяснить данную разницу.
Невозможно также объяснить с помощью такой простой схемы сложную и совершенную регуляцию многих сотен генов. Очевидно, что существуют дополнительные, неизвестные компоненты сигнальной активации с участием Тoll-подобных рецепторов. Вероятно также, что все эти вопросы требуют дальнейшего изучения. Они соприкасаются с одной из наиболее интересных задач генетики, а именно: как относительно небольшим количеством генов, которое имеется у человека и многих других млекопитающих, кодируется огромное количество наблюдаемых фенотипов (фенотипических проявлений) (Benfey, 2008)?
Таким образом, установление функций генов остается одной из насущных задач современной биологии и генетики (Suzuki, 2006). В поисках решения этой проблемы Лаборатория молекулярной генетики врожденного иммунитета ПетрГУ в течение последних лет обратилась к эволюционно разнообразным линиям мышей, которые разошлись с общими предшественниками более миллиона лет назад (Guenet, 2003). С целью номенклатурного различия, эволюционно различные подвиды мышей получили название "диких мышей". В результате многие биологические процессы, включая иммунный ответ, в этих мышах будут активироваться по-другому, в сравнении с классическими лабораторными мышами (Stephan, 2007; Conner, 2008; Conner, 2009; Losick, 2009). Активация перитонеальных макрофагов с помощью ЛПС приводит к значительному повышению уровня TNF уже через 30 минут после активации. В то же время такой ингибитор активации как IL1R-антагонист начинает секретироваться через 24 часа после индукции (Brint, 2004). Как одни и те же клетки способны регулировать процессы активации и секреции, значительно разделенные по времени, пока не известно. Необъяснимыми являются и многие другие процессы. На страницах нашего журнала я призываю обсудить загадки и парадоксы Toll-подобных рецепторов клетки.
Благодарности
Работа выполнена при финансовой поддержке гранта Правительства РФ (Постановление 220), № 11.G34.31.0052 и гранта РФФИ-комфи, № НК 13-04-40267-Н/13.
Библиография
1. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll- like receptor 3. Nature 2001, 413:732-738.
2. Benfey PN, Mitchell-Olds T. From genotype to phenotype: systems biology meets natural variation. Science 2008, 320:495-497.
3. Brint EK, Xu D, Liu H, Dunne A, McKenzie AN, O'Neill LA, Liew FY. ST2 is an inhibitor of interleukin 1 receptor and Toll-like receptor 4 signaling and maintains endotoxin tolerance. Nat Immunol 2004, 5(4):373-379.
4. Conner JR, Smirnova I, Poltorak A. Forward genetic analysis of Toll-like receptor responses in wild-derived mice reveals a novel antiinflammatory role for IRAK1BP1. J Exp Med 2008, 205:305-314.
5. Conner JR, Smirnova I, Poltorak A. A mutation in Irak2c identifies IRAK-2 as a central component of the TLR regulatory network of wild-derived mice. J Exp Med 2009, 206 (7):1615-1631.
6. Guenet JL, Bonhomme F. Wild mice: an ever-increasing contribution to a popular mammalian model. Trends Genet 2003, 19:24-31.
7. Heil F, Ahmad-Nejad P, Hemmi H, Hochrein H, Ampenberger F, Gellert T, Dietrich H, Lipford G, Takeda K, Akira S, Wagner H, Bauer S. The Toll-like receptor 7 (TLR7)-specific stimulus loxoribine uncovers a strong relationship within the TLR7, 8 and 9 subfamily. Eur J Immunol 2003, 33:2987-2997.
9. Janeway CA, Medzhitov R. Introduction: the role of innate immunity in the adaptive immune response. Semin Immunol 1998, 10(5):349-350.
10. Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol 2008, 9:361-368.
11. Kumagai Y, Takeuchi O, Akira S. TLR9 as a key receptor for the recognition of DNA. Adv Drug Deliv Rev 2008, 60:795-804.
12. Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 1996, 86(6):973-983.
13. Losick VP, Stephan K, Smirnova I, Isberg RR, Poltorak A. A hemidominant Naip5 allele in mouse strain MOLF/Ei-derived macrophages restricts Legionella pneumophila intracellular growth. Infect Immun 2009, 77:196-204.
16. Poltorak A, He X, Smirnova I, Liu MY, Huffel CV, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 1998, 282:2085-2088.
17. Smith KD, Andersen-Nissen E, Hayashi F, Strobe K, Bergman MA, Barrett SL, Cookson BT, Aderem A. Toll-like receptor 5 recognizes a conserved site on flagellin required for protofilament formation and bacterial motility. Nat Immunol 2003, 4:1247-1253.
18. Stephan K, Smirnova I, Jacque B, Poltorak A. Genetic analysis of the innate immune responses in wild-derived inbred strains of mice. Eur J Immunol 2007, 37:212-223.
19. Suzuki Y, Roth FP. Systematic genetics swims forward elegantly. Mol Syst Biol 2006, 2:48.
20. Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 1999, 11:443-451.
21. Uematsu S, Sato S, Yamamoto M, Hirotani T, Kato H, Takeshita F, Matsuda M, Coban C, Ishii KJ, Kawai T, Takeuchi O, Akira S. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon- induction. J Exp Med 2005, 201:915-923.
ГОУ ВПО МГУПП НИУВ, Москва
Государственный институт усовершенствования врачей МО РФ, Москва
Роль Toll-подобных рецепторов в патогенезе некоторых дерматозов
Журнал: Клиническая дерматология и венерология. 2011;9(5): 13-20
Сорокина Е.В., Масюкова С.А. Роль Toll-подобных рецепторов в патогенезе некоторых дерматозов. Клиническая дерматология и венерология. 2011;9(5):13-20.
Sorokina EV, Masiukova SA. The role of Toll-like receptors in pathogenesis of certain dermatoses. Klinicheskaya Dermatologiya i Venerologiya. 2011;9(5):13-20. (In Russ.).
ГОУ ВПО МГУПП НИУВ, Москва



В статье рассмотрены сведения об одном из ключевых компонентов иммунной системы - Toll-подобных рецепторах. Обобщены данные исследований функций и структуры Toll-подобных рецепторов и их роли в патогенезе некоторых дерматозов.
ГОУ ВПО МГУПП НИУВ, Москва
Государственный институт усовершенствования врачей МО РФ, Москва
В последние годы появляется все больше сведений, указывающих на роль микробных факторов в реализации аллергопатологии у лиц, генетически к ней предрасположенных. Известно, что микробная инфекция может оказывать в одних случаях предупреждающее, а в других — провоцирующее действие в патогенезе аллергической патологии. Изучение формирования ответа врожденной иммунной системой и ее способности активировать адаптивные механизмы поможет распознать тактику микроорганизмов, позволяющую ускользать от иммунологического надзора.
Исследования TLRs при кожных заболеваниях носят фрагментарный характер. В зарубежной литературе [4—10] имеются единичные статьи обзорного характера, посвященные исследованиям TLRs и цитокинов при некоторых дерматозах, а также изучению функции плазмацитоидных дендритных клеток при аутоиммунной патологии и онкологических заболеваниях — актиническом кератозе, меланоме, базально-клеточной карциноме. Большинство работ посвящено изучению роли TLRs при инфекционных заболеваниях, в том числе дерматозах инфекционной этиологии [6, 11]. Узнавание PAMPs Toll-подобными рецепторами инициирует сигнальный каскад, который вызывает выработку провоспалительных цитокинов, хемокинов, антимикробных пептидов и энзимов в коже, активацию транскрипционных факторов, активатора протеина-1 и ядерного фактора-κB (NF-κB) [12]. TLRs играют ключевую роль в детекции вторгающихся патогенов в коже и инициируют кожный иммунный ответ. В эпидермисе эти рецепторы экспрессируются, в том числе на кератиноцитах и клетках Лангерганса. TLRs идентифицированы на эпителиальных клетках, что указывает на их участие в формировании эпителиального антимикробного барьера [13, 14].
Ряд исследований [15], посвященных изучению роли TLRs при аллергодерматозах, проводился на примере атопического дерматита (АтД). Значительное влияние на течение АтД оказывает колонизация кожи больных патогенными и условно-патогенными микроорганизмами. Часто течение заболевания осложняется инфекциями, вызванными S. aureus или Herpes simplex virus. При АтД при колонизации кожи S. aureus пептидогликан клеточной стенки микроба индуцирует продукцию кератиноцитами в очагах поражения медиаторов воспаления и цитокинов, включая GM-CSF. В ходе изучения экспрессии TLRs в эпидермисе и культуре кератиноцитов установлено, что TLR2 является специфическим рецептором для S. aureus. Дисфункция в TLR2-сигналах является причиной стафилококковой персистенции у пациентов с аллергодерматозами в результате нарушения индукции антимикробных пептидов на примере β-дефензина-2. Подобно другим аллергическим заболеваниям, патофизиология АтД вовлекает Th2-тип иммунного ответа в коже. Когда активируются Th2-клетки, продукция провоспалительных цитокинов и хемокинов поддерживает персистенцию воспаления. При АтД цитокины — интерлейкин-5 (ИЛ-5), ИЛ-13 и фактор некроза опухоли-α (ФНО-α), ИЛ-17, а также ИЛ-31, который экспрессируется прежде всего в коже в Th2-клетках, возможно, играют ведущую роль в развитии воспаления, следующим за кожной экспозицией антигенов [16]. У пациентов, страдающих одновременно АтД и герпетической экземой, наблюдается значительно более низкая продукция антимикробных пептидов, особенно семейства кателицидинов, в кератиноцитах по сравнению с неосложненными формами АтД, а у кателицидин-дефицитных мышей показан высокий уровень репликации вируса простого герпеса 2-го типа [17, 18]. Кателицидины ингибируют функцию гиалуронана — полимера гликозаминогликанов, являющегося эндогенным лигандом для TLR4 и TLR2, и оказывают противовоспалительную активность in vitro и in vivo, способствуя уменьшению высвобождения цитокинов в макрофагах, а дефицит кателицидинов способствует хронизации аллергического дерматита [19]. Мутации в гене R753Q TLR-2 коррелировали с наиболее тяжелым течением АтД, высокими уровнями IgE и большей подверженностью колонизации золотистым стафилококком [18]. Идентифицирован полиморфизм в TLRs у пациентов с АтД. Выявлена ассоциация полиморфизма в TLR2 (R753Q) с тяжелой инфекцией, вызванной S. aureus. У больных АтД обнаружено значительное ослабление в TLR2 опосредованной продукции провоспалительных цитокинов моноцитами периферической крови [20—22]. Ослабление функции TLRs при АтД может способствовать повышению чувствительности к бактериальной и вирусной инфекции, распространению высыпаний и тем самым участвовать в патогенезе заболевания [23]. При сравнительном анализе уровней антимикробных пептидов в здоровой коже при АтД и псориазе в очагах АтД наблюдались более низкие уровни экспрессии, особенно была снижена продукция двух антимикробных пептидов — LL-37 и β-дефензина-2. У больных АтД также наблюдалось снижение экспрессии дефензинов hBD-2 и hBD-3, а Th2-цитокины ИЛ-4 и ИЛ-13 супрессировали индукцию hBD-2 и hBD-3 mRNA туморнекротическим фактором ФНО-α в кератиноцитах [24, 25]. Гипотетически дисфункция в TLR2-сигналах у пациентов с АтД может являться причиной рецидивов стафилококковой инфекции. Несмотря на то, что точная роль TLRs в патофизиологии АтД не окончательно ясна и находится в стадии изучения, значение дисфункции TLR2 в патогенезе этого дерматоза очевидно [21, 25, 26]. Активация же TLR2-сигналов способствовала индукции β-дефензина, способствуя нормализации уровней антимикробных пептидов. Показано, что применение агонистов TLR2 при АтД может напрямую стимулировать Т-клетки, NK-клетки и дендритные клетки к продукции ИФН-γ. Потеря этого эффекта у TLR2–/–мышей может способствовать их нежелательному Th1-ответу кожной чувствительности. При длительном хроническом течении АтД в очагах наблюдается гиперэкспрессия ИФН-γ и это, по мнению авторов, может свидетельствовать о его важной роли в апоптозе кератиноцитов и пролонгировании кожного воспаления [27]. M. Howell и соавт. [28] показали повышение экспрессии гена ИЛ-10 в очагах АтД. Применение при лечении АтД кожных эксплантов с анти-ИЛ-10 стимулировало экспрессию антимикробных пептидов дефензина hBD-2 и кателицидина LL-37.
Определенную роль в патогенезе дерматозов играет микробный фактор, осложняя течение заболевания. Липофильные грибы Malassezia furfur задействованы в развитии псориатических поражений волосистой части головы. Человеческие кератиноциты, инфицированные M. furfur, регулируют TLR2, MyD88, HBD-2, HBD-3 и ИЛ-8 mRNA, повышая экспрессию генов HBD-2 и ИЛ-8. Ингибирование экспрессии генов HBD-2 и ИЛ-8 осуществляется анти-TLR2 нейтрализующими антителами [29]. Хорошо известно, что Propionibacterium acne является ключевой бактерией в развитии акне. Пептидогликан грамположительной бактерии P. acne является экзогенным лигандом для TLR2 и действует как один из сигнальных корецепторов для CD14. J. Kim и соавт. [30], исследовавшие биопсийный материал, полученный из очагов у больных акне, выявили, что TLR2 обильно экспрессировался на перифолликулярных и перибульбарных макрофагах, и концентрация TLR2-несущих клеток и экспрессия in vivo TLR2 и TLR4 в эпидермисе повышалась с эволюцией высыпаний. Грамположительный микроб P. acnes, индуцируя секрецию провоспалительных цитокинов ИЛ-6, ИЛ-8, ИЛ-12, способствует воспалению при акне, а воспалительный ответ индуцируется TLR2. В последних исследованиях выявлено, что ретиноиды, используемые для терапии акне, могут снижать TLR2-сигналы на моноцитах и уменьшают воспалительную реакцию в очагах.
При изучении патогенеза Лайм-боррелиоза было установлено, что липопротеины боррелий, в частности OspA, являются потенциальными активаторами воспалительной реакции за счет связывания с CD14 и TLR2 на макрофагах. В результате этого происходит индукция секреции макрофаг-опосредованных провоспалительных цитокинов рецепторами TLR2, TLR6, TLR1/2, TLR5, TLR9. Димеры рецепторов TLR2/TLR6, TLR2/TLR1 через распознавание триацилированых липопротеинов, таких, как Borrelia burgdorferi OspA, флагеллина, пептидогликанов и зимозана, участвуют в активации ядерного транскрипционного фактора Nf-kB. J. Salazar и соавт. [31], исследовавшие периферическую кровь и аспираты у пациентов с мигрирующей эритемой Афцелиуса-Липшутца, обнаружили высокую экспрессию TLR2, TLR1 и TLR4 на поверхности моноцитов, макрофагов, моноцитоидных и плазмоцитоидных дендритных клеток. Также было установлено, что при развитии мигрирующей эритемы Афцелиуса—Липшутца продукция флагеллина способствует выраженной экспрессии TLR5 и усилению функциональной активности TLR5 на человеческих кератиноцитах. В развитии Лайм-артрита ведущую роль традиционно приписывают Th1-цитокинам, таким как ИФН-γ [32]. Однако в ходе исследований было выявлено, что OspA может участвовать в запуске аутоиммунного воспаления и провоцировать развитие артрита через дифференциацию Th-клеток, которые экспрессируют ИЛ-17 [33]. В эксперименте на B. burgdorferi-вакцинированных мышах также было показано, что ИЛ-17 играет ключевую роль в развитии артрита. Более того, ИЛ-17 может стимулировать фибробласты и синовиоциты к продукции провоспалительных цитокинов, включая ИЛ-6. Данный цитокин в свою очередь может стимулировать дополнительную TGF-β-опосредованную Th17-клеточную пролиферацию до тех пор, пока концентрация инфекционного агента не снизится до значений, не эффективных для индуцирования дальнейшего воспаления [5].
Кератиноциты в неповрежденной коже служат барьерными клетками, не относящимися к иммунной системе. Однако под влиянием повреждения и действия микроорганизмов и их продуктов, а затем цитокинов, они активизируются, экспрессируют молекулы адгезии и начинают выделять цитокины, служащие пусковыми факторами и медиаторами иммунных реакций в коже [34]. Чрезмерная активация Th2-клеток, поддерживаемая TLRs, может привести к перевесу провоспалительных цитокинов и инициации развития хронического воспаления и аутоиммунных заболеваний. При изучении экспрессии TLRs при псориазе результаты носят противоречивый характер. В отличие от АтД, псориаз ассоциируется с Th1-цитокиновым профилем и с Th17-цитокиновым профилем [35, 36]. При исследовании биопсийных фрагментов неповрежденной кожи у больных псориазом и нормальной неизмененной кожи здоровых доноров продемонстрировано, что нормальные кератиноциты экспрессируют TLR1, TLR2 и TLR5 во всех слоях эпидермиса, но более выраженная экспрессия наблюдается в базальных кератиноцитах. В псориатических очагах экспрессия TLR2 была значительно выше в верхних слоях эпидермиса, а экспрессия TLR5 снижена в базальных кератиноцитах [37]. Однако в ряде исследований показано, что базальные кератиноциты в псориатических очагах диффузно и в больших количествах экспрессируют TLR1, TLR2, TLR4, TLR5 и TLR9 [4, 38—41].
Важную роль в реализации врожденного иммунитета наряду с цитокинами играет семейство антимикробных пептидов, которые служат первичной защитой от патогенов, действуя как эндогенные природные антибиотики, выполняя функцию киллинга микробов, и как сигнальные молекулы, вызывающие активацию иммунных клеток. К ним относят дефензины, кателицидины и дермцидин. Однако кожа человека экспрессирует много молекул — ингибиторов протеаз, хемокинов, нейропептидов, обладающих высокой антимикробной активностью.
Проведено исследование уровней антимикробных пептидов у пациентов с АтД и псориазом. Установлено, что у больных АтД в коже наблюдается низкий уровень экспрессии антимикробных пептидов. Известно, что псориатические высыпания, в отличие от АтД, высокорезистентны к суперинфекции патогенными бактериями, такими как S. aureus [3]. Это частично объясняется выявленными у больных псориазом высокой экспрессией и активностью антимикробных пептидов, в том числе кателицидина LL-37 пептида, изолированного из очагов поражения и образующего комплексы с человеческой ДНК, активирующего плазмоцитоидные дендритные клетки [16, 42, 43]. LL-37/self-DNA комплексы в псориатических высыпаниях способствуют повышению продукции ИФН-α вследствие активации TLR9 на плазмоцитоидных дендритных клетках, что впоследствии активирует Т-клеточный ответ, который, вероятно, может лежать в основе кожного воспаления при псориазе [25, 44, 45]. Кроме того, выявлена высокая экспрессия кератиноцитами в псориатических высыпаниях белков теплового шока, которые могут стимулировать секрецию TLR4 на антигенпрезентирующих клетках, главным образом, клетках Лангерганса, играя главную роль в созревании и секреции ФНО-α и ИЛ-12 и участвуя таким образом в иммунопатологии псориаза [39]. L. Miller и соавт. [40, 46] продемонстрировали, что в псориатических высыпаниях и заживающих ранах уровень ростового фактора TGF-α кератиноцитов был повышенным, что приводило к усилению экспрессии TLR5 и TLR9 и TLR-зависимой продукции провоспалительных цитокинов, в том числе ИЛ-8 и β-дефензинов. Однако в ряде исследований [28, 47-49] показано, что при псориазе супрессия реакций гиперчувствительности может быть вызвана нарушением созревания дендритных клеток и снижением высвобождения провоспалительных цитокинов в дендритных клетках вследствие ингибирования функций TLR4 высокими уровнями кателицидина LL-37 в очагах.
Таким образом, накапливаются данные о том, что TLRs и их лиганды не только обеспечивают противоинфекционную защиту, но, способствуя развитию воспалительного ответа, играют важную роль в патогенезе аутоиммунных заболеваний. В ряде исследований показано, что микробные TLR-лиганды провоцируют развитие аутоиммунного заболевания в экспериментальной модели артрита, аллергического энцефаломиелита, миокардита, диабета и атеросклероза. При псориазе наблюдаются высокая экспрессия и активность антимикробных пептидов, что ингибирует функцию TLR4 на дендритных клетках и ведет к нарушению созревания дендритных клеток и высвобождения провоспалительных цитокинов, супрессируя тем самым реакции гиперчувствительности и подавляя воспаление. Высокая экспрессия антимикробных пептидов опосредованно через TLR9 вызывает высвобождение ИФН-α из дендритных клеток, что способствует продукции Th1-профиля цитокинов и, вероятно, поддерживает кожное воспаление при псориазе. Высокая экспрессия белков теплового шока также вызывает повышение секреции TLR4, главным образом на клетках Лангерганса, и как следствие — повышение секреции ФНО-α и ИЛ-12, участвуя в иммунопатологии псориаза. Таким образом, повышенная экспрессия TLR4 и TLR9 играет важную роль в иммунопатогенезе псориаза. Однако известно, что псориатические высыпания, в отличие от АтД, высокорезистентны к суперинфекции патогенными бактериями, такими как S. aureus. Это частично объясняется выявленной у больных псориазом высокой экспрессией и активностью антимикробных пептидов, в том числе кателицидина LL-37 пептида, изолированного из очагов поражения и образующего комплексы с человеческой ДНК, активирующего плазмацитоидные дендритные клетки.
Таким образом, в отличие от псориаза и дерматозов инфекционной этиологии, при АтД наблюдается экспрессия Th2-профиля цитокинов. При аллергодерматозах в ходе исследования роли TLRs на примере АтД выявлены дисфункция в TLR2-сигналах, мутация в TLR2 генах, что приводило к ослаблению TLR2-медиированной продукции провоспалительных цитокинов и коррелировало с тяжелым течением АтД, высокими уровнями IgE и колонизацией S. aureus, развитием пиодермий и инфекции, вызванной вирусом простого герпеса 1-го и 2-го типов. В отличие от псориаза и дерматозов инфекционной этиологии, у пациентов с АтД в коже наблюдаются низкие уровни экспрессии антимикробных пептидов, особенно LL-37 и β-дефензина-2, что также способствует развитию инфекции вируса простого герпеса и колонизации S. aureus. У больных АтД выявлено значительное ослабление в TLR2-опосредованной продукции провоспалительных цитокинов моноцитами периферической крови. Поэтому очевидно, что восстановление функций TLR2 и активация TLR2-сигналов приводит к индукции синтеза антимикробных пептидов у больных аллергодерматозами и способствует профилактике осложнений бактериальной и вирусной природы.
Очевидно, что индукция сигналов через TLRs может обеспечивать не только защиту организма от различных инфекций. Нарушение проводимости данных сигналов приводит к развитию ряда патологических процессов в организме. Чрезмерная продукция провоспалительных цитокинов эндогенными лигандами может стать причиной развития хронического воспаления, аутоиммунных заболеваний. TLRs вовлечены в патогенез всех описанных кожных болезней, таких как Лайм-боррелиоз, псориаз, АтД и акне. Однако эти данные не систематизированы, не дают полного представления о состоянии иммунной системы при указанных патологиях и нуждаются в дальнейшем и более глубоком изучении. Определение роли TLRs при дерматозах целесообразно и, возможно, в перспективе позволит не только прогнозировать течение заболевания, но и поможет оценить и повысить эффективность лечения.
Читайте также: